本文主要是介绍biostar handbook(四)|生物数据及其下载和基本操作,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
2017/11/9 第一版: 生物数据库,基本数据类型(genbank, fasta/fastq),数据上传站点
2017/11/12 第二版:如何利用esearch, efecth快速获取SRR序列号
生物数据库
目前绝大部分数据由NCBI, EMBL-EBI, DDBJ三大机构托管,可划分为五类:
(表格数据来源于INSDC)
| Data type | DDBJ | EMBL-EBI | NCBI |
|---|---|---|---|
| Next generation reads | Sequence Read Archive | European NucleotideArchive (ENA) | Sequence Read Archive |
| Capillary reads | Trace Archive | 同上 | Trace Archive |
| Annotated sequences | DDBJ | 同上 | GenBank |
| Samples | BioSample | 同上 | BioSample |
| Studies | BioProject | 同上 | BioProject |
大部分所需要的数据可在如下站点进行搜索
- GenBank: 存放所有注释和已被发现的DNA序列信息
- SRA: 存放高通量测序产生的短读数据
- PDB: 蛋白3D结构数据库
- uniprot: 最权威的蛋白序列数据库
- UCSC Genome Browser:脊椎动物相关数据库
- FlyBase: 果蝇相关数据库
- WornBase: 蠕虫相关数据库
- SGD: 酵母相关数据库
- RNA-Central: RNA相关资源汇总数据库
- TAIR: 拟南芥相关数据库
- EcoCyc: 大肠杆菌数据库
- 待补充
常见数据格式
GenBank格式
目前测序数据最常储存的格式为GenBank, FASTA和 FASTQ。前两者经常表示策划的序列信息(curated sequence information)。FASTQ格式表示的是从测序仪器中获得的实验数据。
GenBank是最古老的生物信息数据,最早是用来搭建人类可读和机器高效利用之间的桥梁,格式称为固定宽度格式,前10个字符为标识,后面则是相应信息:
LOCUS AF086833 18959 bp cRNA linear VRL 13-FEB-2012
DEFINITION Ebola virus - Mayinga, Zaire, 1976, complete genome.
ACCESSION AF086833
VERSION AF086833.2 GI:10141003
KEYWORDS .
SOURCE Ebola virus - Mayinga, Zaire, 1976 (EBOV-May)ORGANISM Ebola virus - Mayinga, Zaire, 1976Viruses; ssRNA viruses; ssRNA negative-strand viruses;Mononegavirales; Filoviridae; Ebolavirus.
REFERENCE 1 (bases 1 to 18959)AUTHORS Bukreyev,A.A., Volchkov,V.E., Blinov,V.M. and Netesov,S.V.TITLE The VP35 and VP40 proteins of filoviruses. Homology between Marburgand Ebola virusesJOURNAL FEBS Lett. 322 (1), 41-46 (1993)PUBMED 8482365
REFERENCE 2 (bases 1 to 18959)AUTHORS Volchkov,V.E., Becker,S., Volchkova,V.A., Ternovoj,V.A.,Kotov,A.N., Netesov,S.V. and Klenk,H.D.TITLE GP mRNA of Ebola virus is edited by the Ebola virus polymerase andby T7 and vaccinia virus polymerases
GenBank格式的优势在于可读,但是对于数据分析而言不太合适,ReadSeq 可以将将GenBank格式转换为比较简单的版本。
RefSeq,The NCBI Reference Sequence,项目提供了大多数生物的序列记录和相应的信息,为医学,功能和比较分析提供了基准线。

FASTA格式
定义:第一行为>开头,表示为fasta记录开始,随后紧跟序列
问题: 缺乏定义,过于简单
注意:
- 序列行不应太长,
- 不同工具对序列行中的超出字符集(核酸ATCG或蛋白质20种)处理不同
- 序列行如果有多行,除最后一行,前几行应该是等宽的
- 使用大写字母。不同工具会有大小写敏感。如有些工具会认为小写字母是非重复,大写字母象征着重复区域。
- 不同机构对>后面的结构有各自的定义
小写字母可能表示重复区,但是目前还是很难判断哪些是重复区。
注lastz默认过滤小写字母(重复区)
FASTQ格式
FASTQ可以算是FASTA的增加碱基质量版本,很久之前是单行表示所有数据,目前是通过4行进行表示,形如:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
其中第四行是描述碱基质量,通常可以按照如下这样认为
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHI
| | | | | | | | |
0....5...10...15...20...25...30...35...40
| | | | | | | | |
worst................................best
注:
- 目前有许多中FASTQ质量编码版本:有+33, +64两类。以!为0的是,+33,以@开头的是+64
- FASTQ开头的定义也有多个版本见维基百科
FATA/Q 格式操作
为了更好的熟悉FASTA/Q,最好的的方法就是用SeqKit对他们进行一波处理,一探究竟。
# 安装软件
conda install seqkit # for linux
brew install seqkit # for mac
# 下载数据
wget http://data.biostarhandbook.com/reads/duplicated-reads.fq.gz
wget ftp://ftp.ncbi.nih.gov/refseq/release/viral/viral.1.1.genomic.fna.gz
wget ftp://ftp.ncbi.nih.gov/refseq/release/viral/viral.1.protein.faa.gz
我们用seqkit解决如下问题:
- 了解概况
$ seqkit stat *.gz
file format type num_seqs sum_len min_len avg_len max_len
duplicated-reads.fq.gz FASTQ DNA 15,000 1,515,000 101 101 101
viral.1.1.genomic.fna.gz FASTA DNA 8,557 212,624,336 200 24,848 2,473,870
viral.1.protein.faa.gz FASTA Protein 253,637 64,758,934 5 255.3 8,960
- GC含量
seqkit fx2tab --name --only-id --gc viral.1.*fna.gz | head -n 1
NC_021865.1 40.94
- 获取子集, 抽样
# 随机获取id
seqkit sample --proportion 0.001 duplicated-reads.fq.gz | seqkit seq --only-id --name > id.txt
# 根据ID获取序列
seqkit grep --pattern-file id.txt duplicated-reads.fq.gz > duplicated-reads.subset.fq
官方列举出更多的用法,这里不一一赘述, 详见http://bioinf.shenwei.me/seqkit/
自动化数据下载
这里的自动化意味着不经由浏览器查找数据,而是利用命令行根据物种名或者项目号解析下载链接并获取相关数据的过程。
数据下载
所谓“巧妇难为无米之炊”,在数据分析之前得要保证要有数据。 绝大部分的数据,如参考基因组,高通量测序结果可以在如下网站获取
- NCBI web: https://www.ncbi.nlm.nih.gov/
- NCBI FTP: ftp://ftp.ncbi.nlm.nih.gov/
- Ensembl web: http://useast.ensembl.org/index.html
- Ensembl FTP: ftp://ftp.ensembl.org/pub/release-86/
- Biomart: http://www.ensembl.org/biomart/martview/
- UCSC Downloads: http://hgdownload.cse.ucsc.edu/downloads.html
- UCSC FTP: ftp://hgdownload.cse.ucsc.edu/goldenPath/
下载SRA数据
当然目前最常接触的数据是二代测序数据。随着测序价格下降,大家动不动就来一波高通量测序,然后为了可重复就需要把数据上传到专门的服务器。NCBI为了管理这些数据,就专门搞了Short Read Archive,还专门搞出了一个SRA格式用于压缩数据,当然这个SRA格式被吐槽的很惨。这个项目本身的组织方式还是非常棒的
- NCBI BioProject: 以PRJN开头,例如PRJNA257197,通常是一个研究项目的介绍
- NCBI BioSample: 以SAMN/SRS开头, 通常是介绍材料的来源
- SRA Experiment: 以SRX开头, 一个特定样本的单独的测序文库
- SRA Run: 以SRR/ERR开头,只有这个才真正存放数据,只有之中序列号才能用于下载。
给定一个ID, 就可以在https://www.ncbi.nlm.nih.gov/sra或https://www.ebi.ac.uk/ena进行检索,然后用SRAtoolkit或者是curl进行下载。
SRAtoolkit下载的数据来自于NCBI,需要进一步用fastq-dump解压缩。如果不想解压缩的话,就去EBI里进行检索,这里面的数据仅仅经过gzip压缩,下载后直接可以分析。
举个例子,比如说我拿到了一个项目号SRP113349, 我先去NCBI的SRA中进行检索,得到如下子结果。
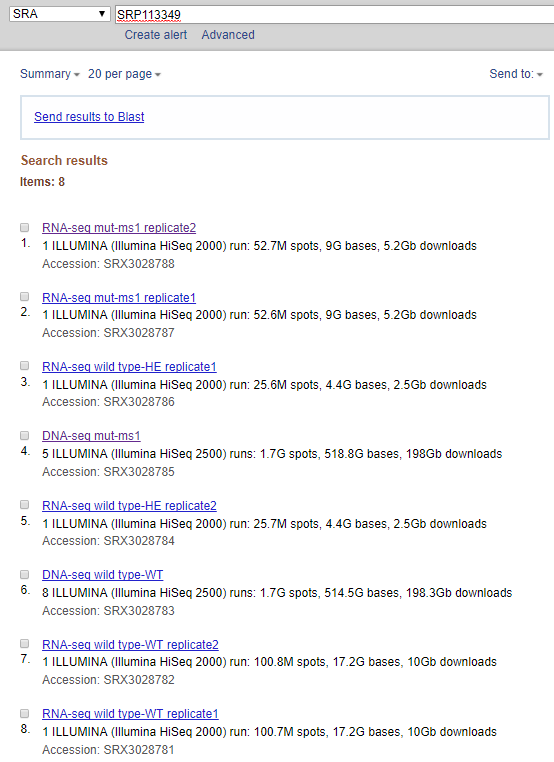
为了获得SRR序列号,我根据需求选择了第一个,并且找到了目标序列号SRR5860104
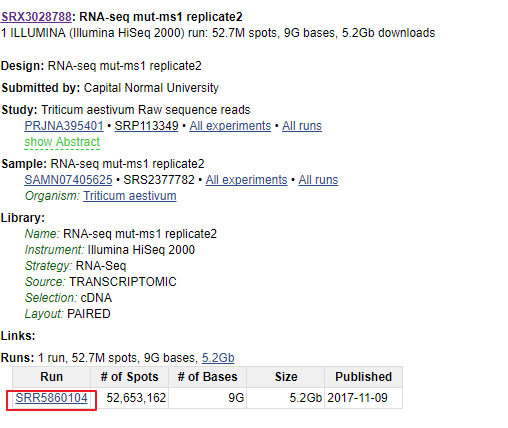
可以直接用sratoolkit的prefetch进行下载。
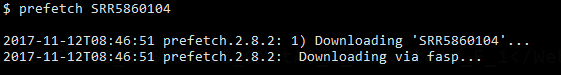
既然我们只需要得到SRR开头的序列号,其实完全没有必要去打开网页一个个翻查,一点都不geek.
使用Entrez检索NCBI
目前NCBI提供了数据存储,分类以及几乎所有生命科学相关的信息。为了便于管理这些数据,NCBI使用了Entrez作为其主要的文本检索和提取工具,用于搞定文献,DNA,蛋白,基因组,基因变异等信息检索。NCBI为了方便用户使用,开放了专门的API,并且为这个API还做了一个命令行工具,叫做Entrez Direct,安装方法如下
#通用方法
cd ~
/bin/bash
perl -MNet::FTP -e \'$ftp = new Net::FTP("ftp.ncbi.nlm.nih.gov", Passive => 1);$ftp->login; $ftp->binary;$ftp->get("/entrez/entrezdirect/edirect.tar.gz");'
gunzip -c edirect.tar.gz | tar xf -
rm edirect.tar.gzbuiltin exit export PATH=$PATH:$HOME/edirect >& /dev/null || setenv PATH "${PATH}:$HOME/edirect"
./edirect/setup.sh
echo "export PATH=\$PATH:\$HOME/edirect" >> $HOME/.bash_profile
# Mac OS
brew install edirect
# conda
conda install entrez-direct
一共提供了8个命令
- esearch : 给定词条(term)对某一个数据库进行检索
- elink: 在esarch的前提下,找到其他相关数据
- efilter: 对之前的结果进行过滤
- efetch : 按照给定的格式下载数据
- xtract : 将XML转变成表格格式
- einfo : 获取
- epost: 上传数据
- nquire: 向网页或CGI服务器发送URL请求
具体的使用方法见 Entrez Direct: E-utilities on the UNIX Command Line, 提供了详细的实例。这里介绍如何使用esearch根据文章上传的项目获取下载所需的SRR号。
esearch -db sra -query SRP113349
# 结果显示有8个结果, 可以通过efetch获取这个8个结果,保存为CSV
esearch -db sra -query SRP113349 | efetch -format runinfo > info.csv
# CSV 的第一列就是SRR
cat info.csv | cut -d ',' -f 1 | head -n 5
# 结合xargs直接完成所有数据下载
cat info.csv | cut -d ',' -f 1 | xargs -i prefetch {}
从此又降低打开网页的必要性,不需要点击那么多下,直接搞定所有项目数据下载。
数据上传
目前还未涉及到上传数据,仅仅列出不同数据上传对应的网站:
- Sequence Read Archive (SRA)
- Gene Expression Omnibus (GEO)
- Database of Short Genetic Variations (dbSNP)
- Database of Genomic Structural Variations (dbVar)
- Database of Expressed Sequence Tags (dbEST)
- Transcriptome Shotgun Assembly Sequence Database (TSA)
- Whole Genome Shotgun Submissions (WGS)
- Metagenomes
- GenBank
- Genomes
简而言之:高通量测序结果上传SRA,功能基因组学数据如RNA-SEQ,CHIP-SEQ上传GEO,变异特异性数据上传dbSNP,转录本组装结果为TSA,其他可以传到GenBank等
这篇关于biostar handbook(四)|生物数据及其下载和基本操作的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





