本文主要是介绍单细胞分析:多模态 reference mapping (2),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
引言
本文[1]介绍了如何在Seurat软件中将查询数据集与经过注释的参考数据集进行匹配。我们展示了如何将来自不同个体的人类骨髓细胞(Human BMNC)的人类细胞图谱(Human Cell Atlas)数据集,有序地映射到一个统一的参考框架上。
我们之前利用参考映射的方法来标注查询数据集中的细胞标签。在Seurat v4版本中,大幅提高了执行集成任务,包括参考映射的速度和内存效率,并且还新增了将查询细胞投影到之前计算好的UMAP(Uniform Manifold Approximation and Projection,均匀流形近似和投影)可视化界面的功能。
内容
在本示例中,我们将展示如何利用一个已经建立的参考数据集来解读单细胞RNA测序(scRNA-seq)查询:
-
根据参考数据集定义的细胞状态集,对每个查询细胞进行标注。 -
将每个查询细胞投影到之前计算完成的UMAP可视化界面上。 -
估算在CITE-seq参考数据集中测量到的表面蛋白的预测水平。
要运行本示例,请确保安装了Seurat v4,该软件可在CRAN上下载。同时,您还需要安装SeuratDisk包。
library(Seurat)
library(ggplot2)
library(patchwork)
options(SeuratData.repo.use = "http://seurat.nygenome.org")
Example 2:绘制人类骨髓细胞图谱
Data
例如,我们将由人类细胞图谱项目生成的,来自八位不同捐献者的人类骨髓单核细胞(BMNC)数据集进行了映射。我们以之前使用加权最近邻分析(WNN)方法分析过的人类BMNC的CITE-seq参考集作为比对标准。
本文除了展示与之前PBMC案例相同的参考映射功能外,还进一步介绍了:
-
如何构建一个监督的主成分分析(sPCA)转换。 -
如何将多个不同的数据集依次映射到同一个参考集上。 -
采取哪些优化措施来提高映射过程的速度。
# Both datasets are available through SeuratData
library(SeuratData)
#load reference data
InstallData("bmcite")
bm <- LoadData(ds = "bmcite")
#load query data
InstallData('hcabm40k')
hcabm40k <- LoadData(ds = "hcabm40k")
参考数据集构建了一个加权最近邻(WNN)图,该图体现了在本次CITE-seq实验中RNA和蛋白质数据的加权整合情况。
基于这个WNN图,我们可以生成一个UMAP(Uniform Manifold Approximation and Projection)的可视化表示。在计算过程中,我们设置参数return.model为TRUE,这样就可以将待查询的数据集映射到这个UMAP可视化空间中。
bm <- RunUMAP(bm, nn.name = "weighted.nn", reduction.name = "wnn.umap",
reduction.key = "wnnUMAP_", return.model = TRUE)
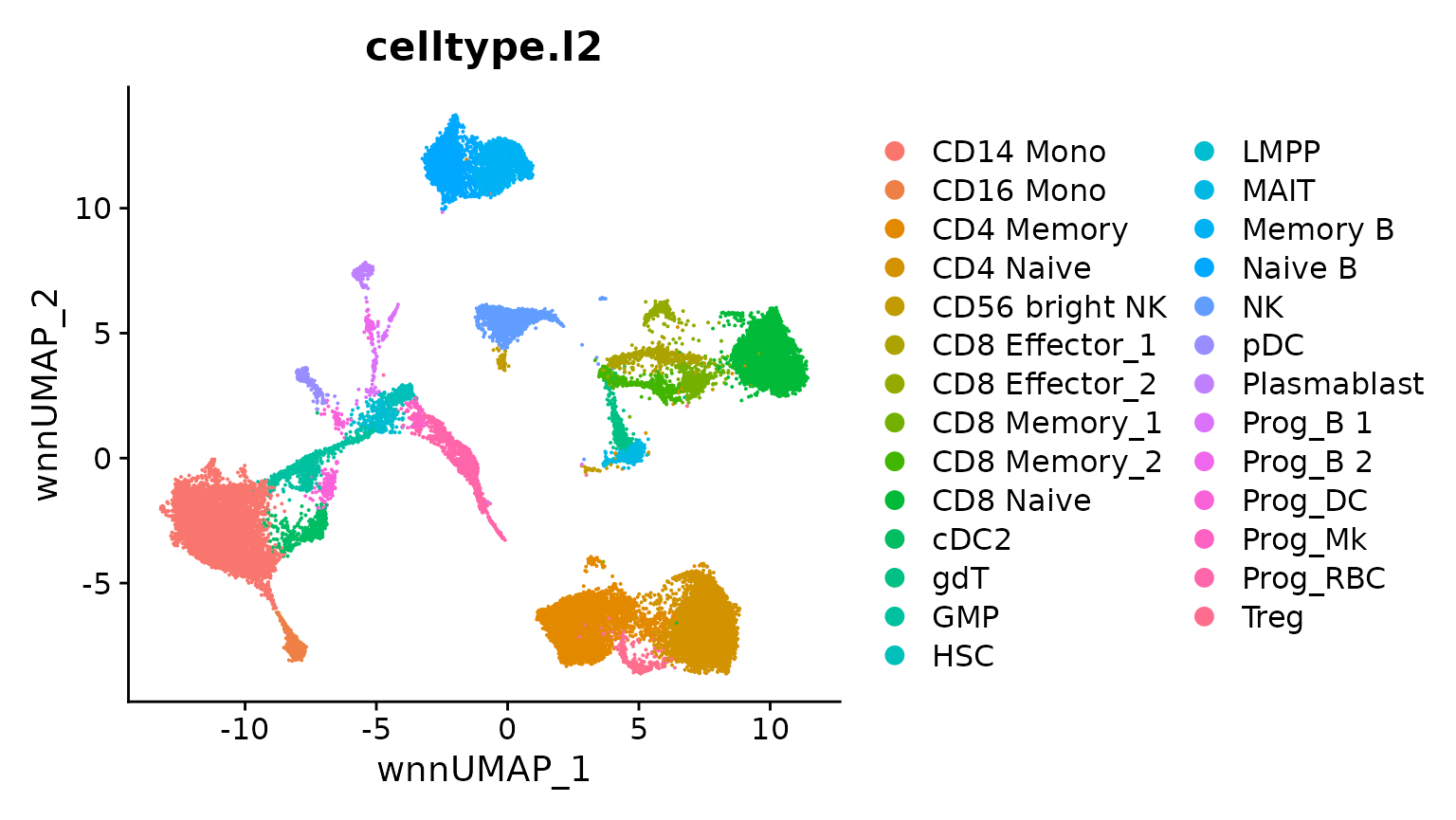
DimPlot(bm, group.by = "celltype.l2", reduction = "wnn.umap")
计算 sPCA 变换
如我们在论文中所述,我们首先执行一个“监督式”的主成分分析(PCA)。该分析旨在找出转录组数据的最佳转换方式,以最准确地反映加权最近邻(WNN)图中的结构特征。通过这种方法,我们可以将蛋白质和RNA的测量值进行加权组合,以“指导”PCA的计算过程,从而凸显出数据中最为重要的变异因素。一旦计算出这种转换,就可以将其应用到任何查询数据集上。尽管我们也可以计算并应用传统的PCA投影,但在处理通过WNN分析构建的多模态参考数据时,我们更推荐使用监督式PCA(sPCA)。
sPCA的计算过程只需进行一次,之后就可以快速地将其应用到每一个查询数据集上。
bm <- ScaleData(bm, assay = 'RNA')
bm <- RunSPCA(bm, assay = 'RNA', graph = 'wsnn')
计算缓存的邻居索引
鉴于我们需要将多个查询样本与同一个参考集进行比对,我们可以对那些仅与参考集相关的特定步骤进行缓存处理。这个步骤虽然是可选的,但在处理多个样本的映射时,它可以有效提升运算速度。
我们首先在参考集的监督式PCA(sPCA)空间内计算出前50个最近邻。然后,我们将这些信息保存在Seurat对象的spca.annoy.neighbors属性中,并通过设置cache.index = TRUE来缓存annoy索引数据结构。
bm <- FindNeighbors(
object = bm,
reduction = "spca",
dims = 1:50,
graph.name = "spca.annoy.neighbors",
k.param = 50,
cache.index = TRUE,
return.neighbor = TRUE,
l2.norm = TRUE
)
-
如何保存和加载缓存的烦恼索引?
如果您需要保存或加载一个利用 "annoy" 方法和启用了缓存索引(通过设置 cache.index = TRUE)创建的 Neighbor 对象的缓存索引,可以使用 SaveAnnoyIndex() 和 LoadAnnoyIndex() 这两个函数来完成。需要注意的是,这个索引不能通过常规方式保存到 RDS 或 RDA 文件,这意味着它不会在 R 会话重新启动或使用 saveRDS/readRDS 函数保存和读取包含该索引的 Seurat 对象时被正确保留。因此,每次当 R 重新启动或者您从 RDS 文件加载参考 Seurat 对象时,都需要使用 LoadAnnoyIndex() 函数来重新将 Annoy 索引加载到 Neighbor 对象中。SaveAnnoyIndex() 函数生成的文件可以与参考 Seurat 对象一起分发,以便在需要时将其添加到参考对象中的 Neighbor 对象里。
bm[["spca.annoy.neighbors"]]
## A Neighbor object containing the 50 nearest neighbors for 30672 cells
SaveAnnoyIndex(object = bm[["spca.annoy.neighbors"]], file = "/brahms/shared/vignette-data/reftmp.idx")
bm[["spca.annoy.neighbors"]] <- LoadAnnoyIndex(object = bm[["spca.annoy.neighbors"]], file = "/brahms/shared/vignette-data/reftmp.idx")
查询数据集预处理
本节我们将展示如何将来自多位捐献者的骨髓样本与一个多模态骨髓参考集进行比对。这些待查询的数据集来源于人类细胞图谱(Human Cell Atlas,HCA)的免疫细胞图谱中的骨髓数据集,可以通过SeuratData包访问。提供的数据集是一个合并后的对象,涵盖了8位捐献者的数据。我们首先需要将这些数据拆分成8个独立的Seurat对象,对应每位捐献者,然后分别进行映射分析。
library(dplyr)
library(SeuratData)
InstallData('hcabm40k')
hcabm40k.batches <- SplitObject(hcabm40k, split.by = "orig.ident")
接下来,我们按照参考数据集的处理方式对查询数据集进行标准化处理。具体来说,参考数据集是通过NormalizeData()函数采用对数标准化的方法进行处理的。如果参考数据集是利用SCTransform()函数进行标准化的,那么查询数据集同样需要应用SCTransform()函数来进行标准化处理。
hcabm40k.batches <- lapply(X = hcabm40k.batches, FUN = NormalizeData, verbose = FALSE)
Mapping
接下来,我们在每位捐献者的数据集与多模态参考集之间确定锚点。为了缩短映射时间,我们采用了一种优化的命令,该命令通过输入预先计算好的参考邻居集合,并关闭锚点筛选功能来实现效率提升。
anchors <- list()
for (i in 1:length(hcabm40k.batches)) {
anchors[[i]] <- FindTransferAnchors(
reference = bm,
query = hcabm40k.batches[[i]],
k.filter = NA,
reference.reduction = "spca",
reference.neighbors = "spca.annoy.neighbors",
dims = 1:50
)
}
然后我们单独映射每个数据集。
for (i in 1:length(hcabm40k.batches)) {
hcabm40k.batches[[i]] <- MapQuery(
anchorset = anchors[[i]],
query = hcabm40k.batches[[i]],
reference = bm,
refdata = list(
celltype = "celltype.l2",
predicted_ADT = "ADT"),
reference.reduction = "spca",
reduction.model = "wnn.umap"
)
}
探索映射结果
现在映射已完成,我们可以可视化各个对象的结果
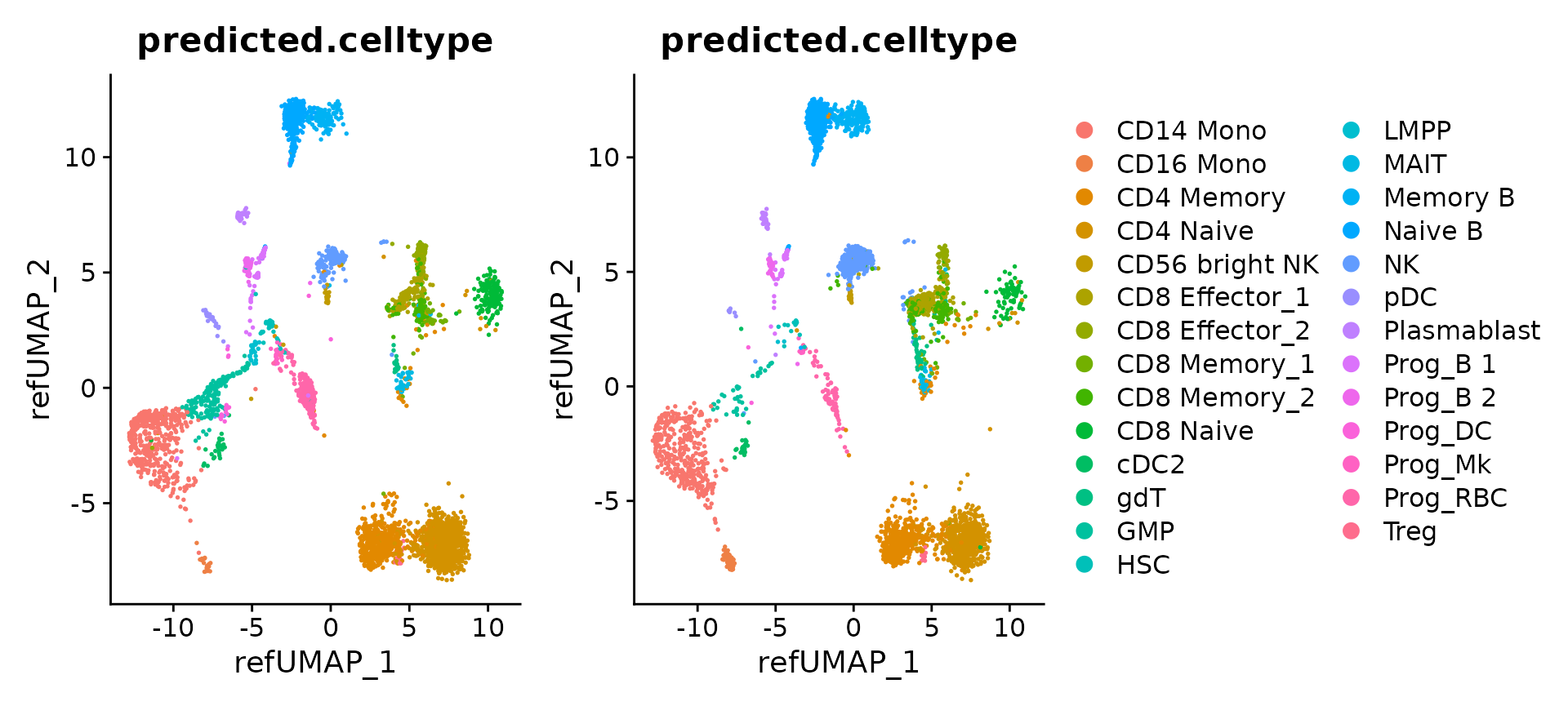
p1 <- DimPlot(hcabm40k.batches[[1]], reduction = 'ref.umap', group.by = 'predicted.celltype', label.size = 3)
p2 <- DimPlot(hcabm40k.batches[[2]], reduction = 'ref.umap', group.by = 'predicted.celltype', label.size = 3)
p1 + p2 + plot_layout(guides = "collect")
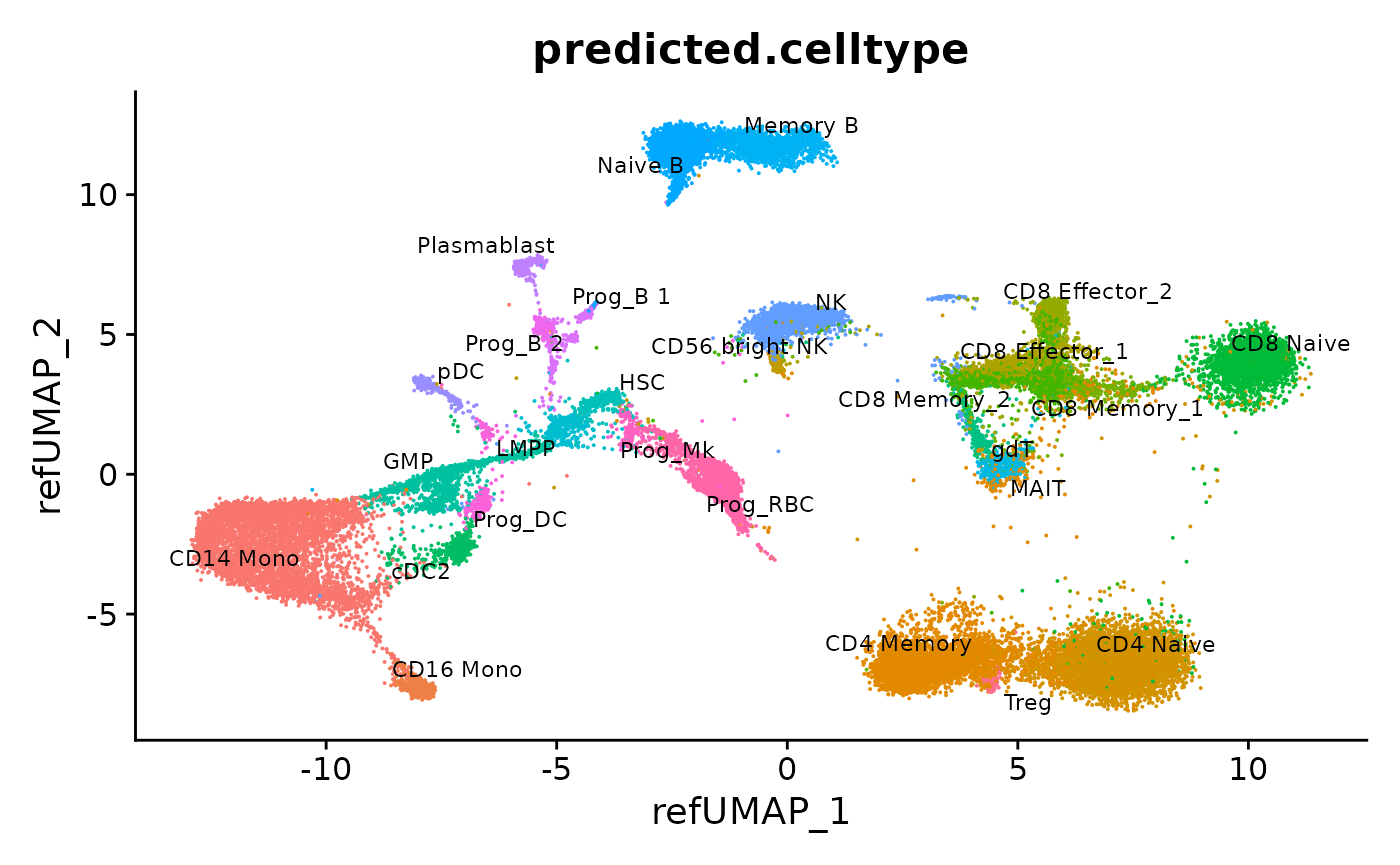
我们还可以把所有的数据对象合并成一个统一的数据集。需要注意的是,这些数据对象都已经通过参考集被整合到了一个共同的分析空间中。之后,我们就能够将这些数据的分析结果一并展现出来。
# Merge the batches
hcabm40k <- merge(hcabm40k.batches[[1]], hcabm40k.batches[2:length(hcabm40k.batches)], merge.dr = "ref.umap")
DimPlot(hcabm40k, reduction = "ref.umap", group.by = "predicted.celltype", label = TRUE, repel = TRUE, label.size = 3) + NoLegend()
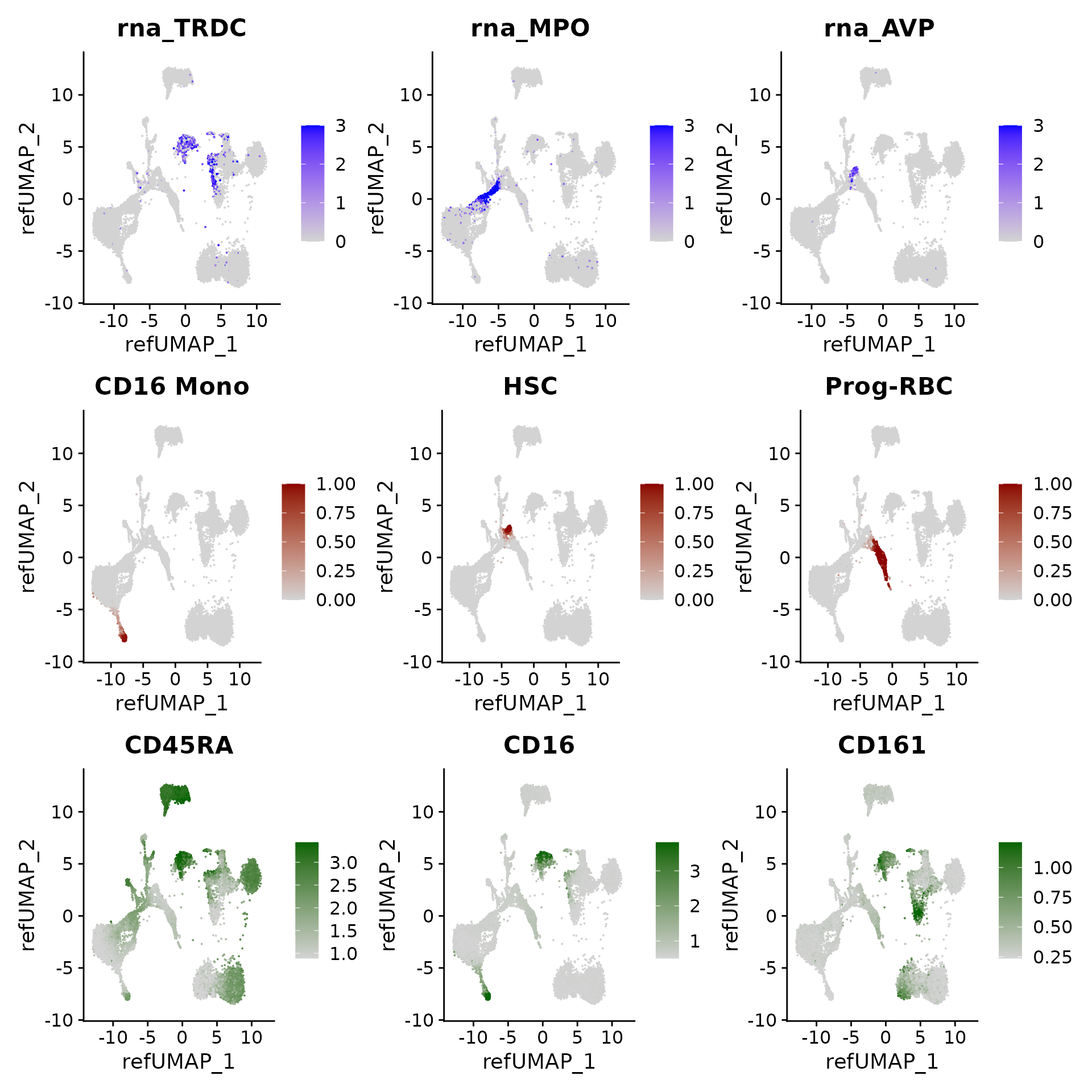
我们可以对查询细胞中的基因表达模式、聚类预测得分以及(估算得到的)表面蛋白水平进行可视化展示:
p3 <- FeaturePlot(hcabm40k, features = c("rna_TRDC", "rna_MPO", "rna_AVP"), reduction = 'ref.umap',
max.cutoff = 3, ncol = 3)
# cell type prediction scores
DefaultAssay(hcabm40k) <- 'prediction.score.celltype'
p4 <- FeaturePlot(hcabm40k, features = c("CD16 Mono", "HSC", "Prog-RBC"), ncol = 3,
cols = c("lightgrey", "darkred"))
# imputed protein levels
DefaultAssay(hcabm40k) <- 'predicted_ADT'
p5 <- FeaturePlot(hcabm40k, features = c("CD45RA", "CD16", "CD161"), reduction = 'ref.umap',
min.cutoff = 'q10', max.cutoff = 'q99', cols = c("lightgrey", "darkgreen") ,
ncol = 3)
p3 / p4 / p5
Source: https://satijalab.org/seurat/articles/multimodal_reference_mapping
本文由 mdnice 多平台发布
这篇关于单细胞分析:多模态 reference mapping (2)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!






