本文主要是介绍从基因组获取fasta文件并计算Nx0脚本,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
从基因组获取fasta文件并计算Nx0脚本
import sys
from Bio import SeqIO
from Bio.SeqRecord import SeqRecord
from Bio.Seq import Seq
import re #正则表达式
我们先导入模块,sys将参数提取到脚本外,实现把路径跟在脚本后面直接跑即可。SeqIO是Bio里面的子包,用于序列信息提取,re为正则表达式载入。
读取文件
fn = 'sys.argv[1]'
seq_index = SeqIO.index(fn,'fasta')
chr_len = {k:len(v.seq) for k,v in seq_index.items()} # 列表推导式 迭代文章有讲 取出id并且算出序列长。
我们一般倾向于seq_index的使用保存为字典后比较好提取内容。
计算
简单信息:总长、条数、平均长度
n = len(chr_len)
total_len = sum(chr_len.values())
mean_len = total_len / n
最长序列
longest_id, longest_len = max(chr_len.items(), key=lambda x:x[1])
使用max函数求最长序列。
N50
def Nx0(l,x):'''该函数用于计算Nx0, 接受两个参数:l : 长度列表x : N50 | N60 | N70...返回两个值:idx : Nx0 的编号nx0 : Nx0 的长度'''l = sorted(l, reverse=True)p = int(x[1:]) / 100total_len = sum(l)nx0 = idx = cumsum = 0for i,v in enumerate(l, start = 1):cumsum += vif cumsum >= total_len * p:idx = inx0 = vbreak return idx, nx0
定义函数,输入两个参数,然后输出两个参数,if的一个简单判断。
N count 和Gaps
n_count =0
gaps = 0
for k,v in seq_index.items():s= str(v.seq) # 正则表达式需要转换为字符串。n_count += s.count('N')gaps += len(re.split('N+',s))-1 #通过切割序列数来定义Gaps。
计算序列碱基里面的N的数量,还有Gaps的数量。
打印
fi_name = fn.split('/')[-1]
print('stats for {}'.format(fi_name))
print('sum = {}, n = {}, ave = {}, largest = {}'\.format(total_len, n, mean_len, longest_id))for n in ('N50', 'N60', 'N70', 'N80', 'N90', 'N100'):idx, nx0 = Nx0(chr_len.values(), n)print('{} = {}, n = {}'.format(n, nx0, idx))print('count ={}'.format(n_count))print('Gaps = {}'.format(gaps))
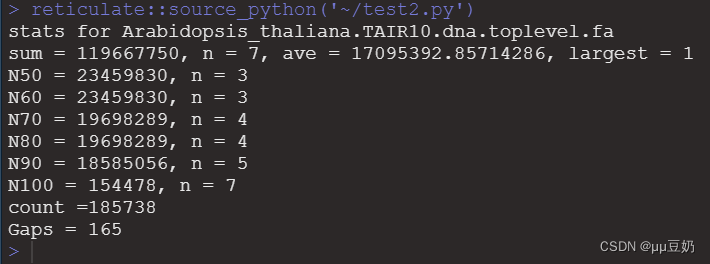
感觉在R语言里面,执行Python脚本感觉还是挺麻烦的,特别是我的终端特别的卡,而且终端输入文件路径时正斜线与反斜线的交叉使用让我很头疼,虽然能联动但是感觉还是挺麻烦的,不知道是我技术有限还是因为确实如此。
小结
弥补了上一个脚本不能计算Count与Gaps的问题,并且学习了新的模块,也不算模块吧,相当于新的包,提取序列的id,name,seq,还是挺方便的,大小写转化,反向互补什么的,就不需要依赖其它网站或者软件了,一行代码就搞定了,这点感觉还是挺不错的。特别是批量转换的时候,会体现出很大的优势。
没什么好聊的了,日常鸡汤:对待生命你不妨大胆冒险一点, 因为好歹你要失去它。如果这世界上真有奇迹,那只是努力的另一个名字。没有人可以拯救自己,到最后拯救你的只有你自己。绝处逢生的喜悦,大难不死的庆幸,都不是你所想象的“人品守恒”。在这个世界面前,生活是无比具体而烦琐的藤蔓,你只有从中体会到酸甜苦辣才知道它最后的余香。生命是需要奋斗的,奋斗与不奋斗,造就的结果截然不同。生无所息,保持奋斗的姿态,让世界变得如此灿烂,让你的人生绚烂多姿。千万不能满足小溪的平缓,否则你也就满足了自己的平庸,只有欣赏到山峰的险峻,才有机会欣赏自己。
这篇关于从基因组获取fasta文件并计算Nx0脚本的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!




