本文主要是介绍ncbi-genome-download批量下载基因组数据,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
1. ncbi-genome-download 的下载和安装
ncbi-genome-download 是一个可以直接从NCBI上批量下载序列的软件,支持下载多种格式
利用 conda 对其直接安装 参考
#创建环境
conda create -n ncbi_genome_download
#激活环境
conda activate ncbi_genome_download
# 安装
conda install -c bioconda ncbi-genome-download
2.常用的参数
-s:选择数据库(genbank,refseq),默认是refseq数据库-F:需要下载基因组的格式,可以多种格式同时下载,用逗号隔开,默认是genbank格式-l:序列组装程度,可以多种格式同时下载,用逗号隔开-g:需要下载序列的属,后面要指定类群,比如bacteriaS:下载的具体的菌种名称,用逗号隔开,也可以写入一个文件中,一行一个菌种名称-o:输出的文件名称-r:失败时重新连接的次数,默认是0次--flat-output:将下载的文件输入到一个目录中,不创建新的子文件(即下载的数据在指定的文件夹中,每个 Taxonomy ID 一个压缩文件)
参数详情:
(ncbi_genome_download) [hgzhong@head01 ~]$ ncbi-genome-download -h
usage: ncbi-genome-download [-h] [-s {refseq,genbank}] [-F FILE_FORMATS] [-l ASSEMBLY_LEVELS] [-g GENERA] [--genus GENERA] [--fuzzy-genus] [-S STRAINS] [-T SPECIES_TAXIDS] [-t TAXIDS] [-A ASSEMBLY_ACCESSIONS] [--fuzzy-accessions] [-R REFSEQ_CATEGORIES] [--refseq-category REFSEQ_CATEGORIES] [-o OUTPUT] [--flat-output] [-H] [-P] [-u URI] [-p N] [-r N] [-m METADATA_TABLE] [-n] [-N] [-v] [-d] [-V] [-M TYPE_MATERIALS] groups positional arguments: groups The NCBI taxonomic groups to download (default: all). A comma-separated list of taxonomic groups is alsopossible. For example: "bacteria,viral"Choose from:['all', 'archaea', 'bacteria', 'fungi','invertebrate', 'metagenomes', 'plant', 'protozoa','vertebrate_mammalian', 'vertebrate_other', 'viral']optional arguments:-h, --help show this help message and exit-s {refseq,genbank}, --section {refseq,genbank}NCBI section to download (default: refseq)-F FILE_FORMATS, --formats FILE_FORMATSWhich formats to download (default: genbank).A comma-separated list of formats is also possible. Forexample: "fasta,assembly-report". Choose from:['genbank', 'fasta', 'rm', 'features', 'gff','protein-fasta', 'genpept', 'wgs', 'cds-fasta', 'rna-fna', 'rna-fasta', 'assembly-report', 'assembly-stats', 'translated-cds', 'all']-l ASSEMBLY_LEVELS, --assembly-levels ASSEMBLY_LEVELSAssembly levels of genomes to download (default: all).A comma-separated list of assembly levels is alsopossible. For example: "complete,chromosome". Choosefrom: ['all', 'complete', 'chromosome', 'scaffold','contig']-g GENERA, --genera GENERAOnly download sequences of the provided genera. Acomma-seperated list of genera is also possible. Forexample: "Streptomyces coelicolor,Escherichia coli".(default: [])--genus GENERA Deprecated alias of --genera--fuzzy-genus Use a fuzzy search on the organism name instead of anexact match.-S STRAINS, --strains STRAINSOnly download sequences of the given strain(s). Acomma-separated list of strain names is possible, aswell as a path to a filename containing one name perline.-T SPECIES_TAXIDS, --species-taxids SPECIES_TAXIDSOnly download sequences of the provided species NCBItaxonomy IDs. A comma-separated list of species taxidsis also possible. For example: "52342,12325".(default: [])-t TAXIDS, --taxids TAXIDSOnly download sequences of the provided NCBI taxonomyIDs. A comma-separated list of taxids is alsopossible. For example: "9606,9685". (default: [])-A ASSEMBLY_ACCESSIONS, --assembly-accessions ASSEMBLY_ACCESSIONSOnly download sequences matching the provided NCBIassembly accession(s). A comma-separated list ofaccessions is possible, as well as a path to afilename containing one accession per line.--fuzzy-accessions Use a fuzzy search on the entry accession instead ofan exact match.-R REFSEQ_CATEGORIES, --refseq-categories REFSEQ_CATEGORIESOnly download sequences of the provided refseqcategories [refrerence, representative, na]. A comma-separated list of categories is also possible.(default: download all categories)--refseq-category REFSEQ_CATEGORIESDeprecated alias for --refseq-categories-o OUTPUT, --output-folder OUTPUTCreate output hierarchy in specified folder (default:/data/home/hgzhong)--flat-output Dump all files right into the output folder withoutcreating any subfolders.-H, --human-readable Create links in human-readable hierarchy (might failon Windows)-P, --progress-bar Create a progress bar for indicating the downloadprogress-u URI, --uri URI NCBI base URI to use (default:https://ftp.ncbi.nih.gov/genomes)-p N, --parallel N Run N downloads in parallel (default: 1)-r N, --retries N Retry download N times when connection to NCBI fails(default: 0)-m METADATA_TABLE, --metadata-table METADATA_TABLESave tab-delimited file with genome metadata-n, --dry-run Only check which files to download, don't downloadgenome files.-N, --no-cache Don't cache the assembly summary file in/data/home/hgzhong/.cache/ncbi-genome-download.-v, --verbose increase output verbosity-d, --debug print debugging information-V, --version print version information-M TYPE_MATERIALS, --type-materials TYPE_MATERIALSSpecifies the relation to type material for theassembly (default: any). "any" will include assemblieswith no relation to type material value defined, "all"will download only assemblies with a defined value. Acomma-separated list of relatons. For example:"reference,synonym". Choose from: ['any', 'all','type', 'reference', 'synonym', 'proxytype','neotype'] .3. 批量下载基因组数据
3.1 根据属名下载
将需要下载的属名放置至一个txt文档(换行),利用参数--genera pant_download.txt plant
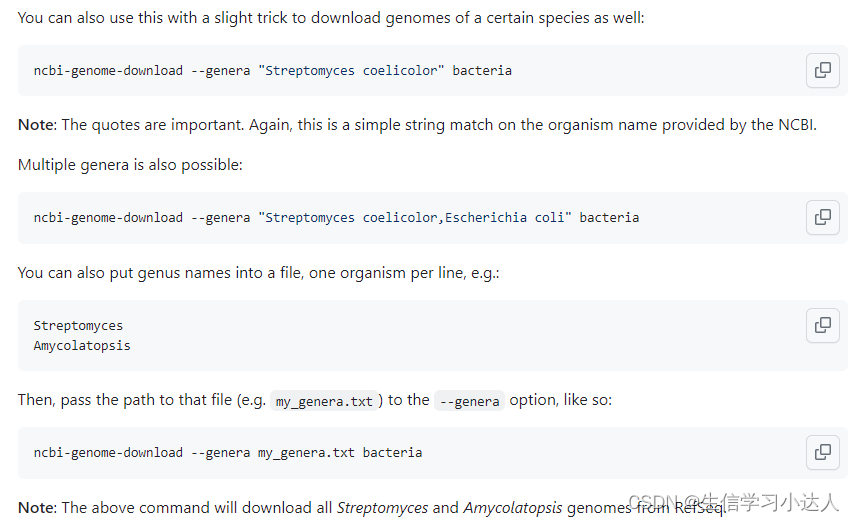
3.2 根据物种的 ID下载
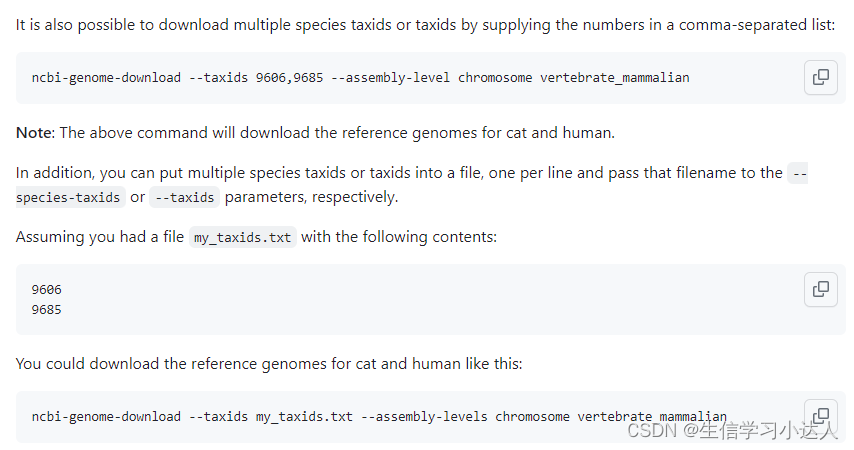
将需要下载的物种的分类 ID 放置至一个txt文档(换行),利用参数--taxids my_taxids.txt
再加上参数 --assembly-levels 指定下载的基因组的不同类型(包括contig,scaffold,chromosome,all,compete)
不同物种的 taxonomy id查询地址 taxonomy id query
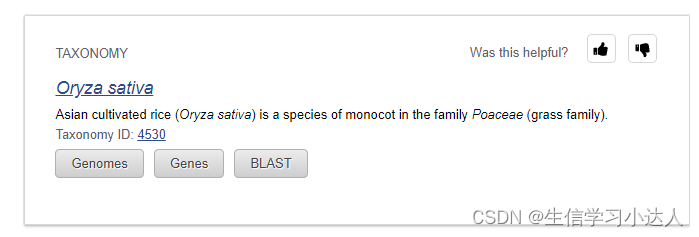
例如 Oryza sativa 的 taxonomy id为4530
3.3 根据物种拉丁名下载
当你有一系列菌种需要下载时,你可以将这一系列菌种名保存到一个txt文件里,每个菌种名为一行,文件名为genera.txt
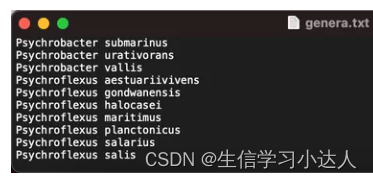
希望下载这些菌种基因组中的cds序列,并将下载的每个文件放在MyGenera文件夹中,在MyGenera目录下进入终端,运行:
ncbi-genome-download --genera genera.txt bacteria --flat-output --formats cds-fasta然后,每个物种均会自动下载好指定的基因组类型序列
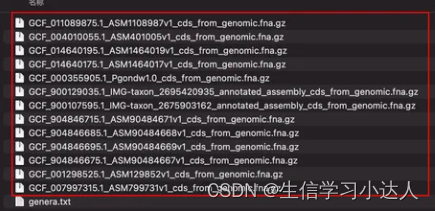
这样genera文本中的所有菌种的基因组cds序列就一条代码下载完成了。
注意:genera.txt文本中有10个菌种名,而下载了13个文件,说明有的菌种名下面有来自不同上传者提供的基因组信息(即一对多)
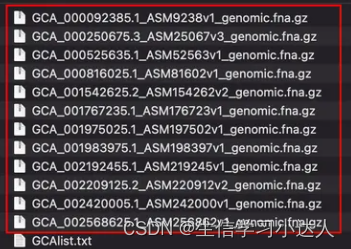
如下图,我要下载GCAlist.txt文本中的基因组序列的fasta文件,并保存在Assembly文件夹中,在Assembly目录下进入终端,运行:
ncbi-genome-download --assembly-accessions GCAlist.txt bacteria --section genbank --for
4.帮助查询
查询地址 帮助文档
- 查看版本
ncbi-genome-download -V- 查看帮助
ncbi-genome-download -h- 语法格式
ncbi-genome-download [optional arguments] groupsoptional arguments为可选参数,详细介绍见下文
groups为物种选择,可选['all', 'archaea', 'bacteria', 'fungi', 'invertebrate', 'metagenomes', 'plant', 'protozoa', 'vertebrate_mammalian', 'vertebrate_other', 'viral'],可选项即为NCBI的FTP下载目录Index of/genomes/refseq和Index of/genomes/genbank下的内容
- 可选参数
--section
指定下载的数据库,可选['refseq', 'genbank'],默认refseq
--formats
指定下载的文件格式,可选['genbank', 'fasta', 'rm', 'features', 'gff', 'protein-fasta', 'genpept', 'wgs', 'cds-fasta', 'rna-fna', 'rna-fasta', 'assembly-report', 'assembly-stats', 'all'],默认genbank
--assembly-levels
指定下载的基因组组装水平,可选['all', 'complete', 'chromosome', 'scaffold', 'contig'],默认all
--genera
根据菌种名下载,后面可接想要下载的菌种名,如--genera 'Rhizobium alamii'
--taxids
根据NCBI taxonomy ID下载,后面可接想要下载的菌种的taxonomy ID,如--taxids '492774'
(还以Rhizobium alamii举例,通过NCBI Taxonomy Browser可以查询到该菌种的txid为492774)
--assembly-accessions
根据assembly accession下载,后面可接想要下载的菌种的assembly accession,如--assembly-accessions ‘GCF_000799895.1’
⚠️注意:因为默认下载的数据库是refseq,所以选择RefSeq assembly accession下载时无需加--section参数即可正常下载,如果要根据GenBank assembly accession下载,请再加上--section genbank。
--output-folder
指定下载目录,后面可接你想要存放的下载目录,如--output-folder ~/Downloads(下载到当前用户的下载文件夹中)
--flat-output
直接将下载的文件放入指定文件夹中,不创建子文件夹
详情参考 ncbi-genome-download工具
5. 核查下载情况
由于利用ncbi-genome-download下载物种的基因组数据时存在未成功下载(后续需自己手动下载)的情况,所有需要将当前目录下的a.genomic.fna.gz文件进行汇总
5.1 利用grep命令查找
随后将sequence_name.txt文件导入excel表中,与自己需要下载的物种进行vlookup函数匹配,找出未成功下载的基因组序列的物种名
grep ".*genomic.fna.gz” ./present dictionary > sequence_name.txt5.2 将.txt文件转换成.bat文件
另一种方法,比较简单实用:

这篇关于ncbi-genome-download批量下载基因组数据的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!





