本文主要是介绍使用purge_haplogs处理基因组杂合区域,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
FALCON和Canu的组装后会得到一个单倍型融合的基因组,用来表示二倍体基因组。之后,FALCON Unzip和Supernova这类软件进一步处理其中等位基因区域,将这部分区间进行拆分。
当基因组某些区域可能有着比较高的杂合度,这会导致基因组该区域的两个单倍型被分别组装成primary contig, 而不是一个为primary contig, 另一个是associated haplotig. 如果下游分析主要关注于单倍型,这就会导致一些问题。
那么有没有解决方案呢?其实也很好办,就是找到相似度很高的contig,将他们拆分。目前已经有一些软件可以完成类似任务,如 HaploMerger2, Redundans, 这不过这些软件主要处理二代组装结果。
purge_haplogs则是最近开发,用于三代组装的基因组。它根据minimap2的比对结果,通过分析比对read的覆盖度决定谁去谁留。该工具适用于单倍型组装软件,例如 Canu, FALCON或 FALCON-Unzip primary contigs, 或者是分相后的二倍体组装(Falcon-Unzip primary contigs + haplotigs 。
它的工作流程如下图所示。一般只需要两个输入文件,组装草图(FASTA格式) 和 比对的BAM文件。同时还可以提供重复序列注释的BED文件,辅助处理高重复的contig。
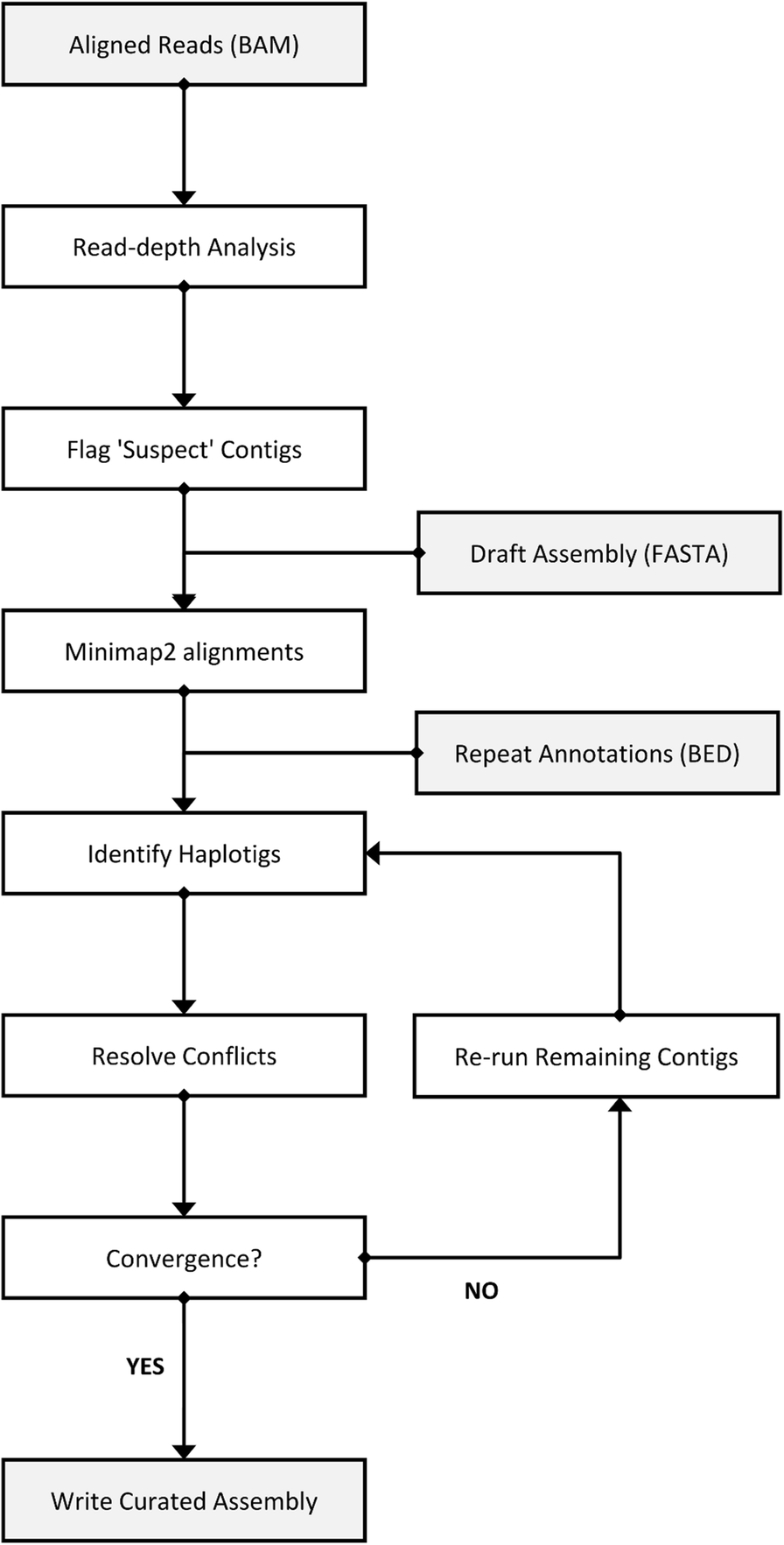
建议: 用原来用于组装的read进行比对。对于多个匹配的read,建议采取random best,也就是随便找一个。
软件安装
purge_haplotigs依赖软件比较多,手动安装会很麻烦,但是他可以直接用bioconda装
conda create -n purge_haplotigs_env
conda activate purge_haplotigs_env
conda install purge_haplotigs
安装完成后需要一步测试
purge_haplotigs test
简明教程
数据准备。 需要下载的数据集分为两个部分,一个是FALCON-Unzip后的primary contig 和 halplotigs. 另一个则是已经比完后的BAM文件
mkdir purge_haplotigs_tutorial
cd purge_haplotigs_tutorial
wget https://zenodo.org/record/841398/files/cns_h_ctg.fasta
wget https://zenodo.org/record/841398/files/cns_p_ctg.aligned.sd.bam # 1.7G
wget https://zenodo.org/record/841398/files/cns_p_ctg.aligned.sd.bam.bai wget https://zenodo.org/record/841398/files/cns_p_ctg.fasta
wget https://zenodo.org/record/841398/files/cns_p_ctg.fasta.fai
当然我们不可能直接就拿到比对好的BAM文件,我们一般是有组装后的基因组以及用于组装的subread,假设这两个文件命名为, genome.fa 和 subreads.fasta.gz.
minimap2 -ax map-pb genome.fa subreads.fasta.gz \| samtools view -hF 256 - \| samtools sort -@ 8 -m 1G -o aligned.bam -T tmp.ali
如果你有二代测序数据,也可以用BWA-MEM进行比对得到BAM文件。
第一步:使用purge_haplotigs readhist从BAM中统计read深度,绘制柱状图。
purge_haplotigs readhist -b aligned.bam -g genome.fasta -t 20
# -t 线程数, 不宜过高,过高反而没有效果。
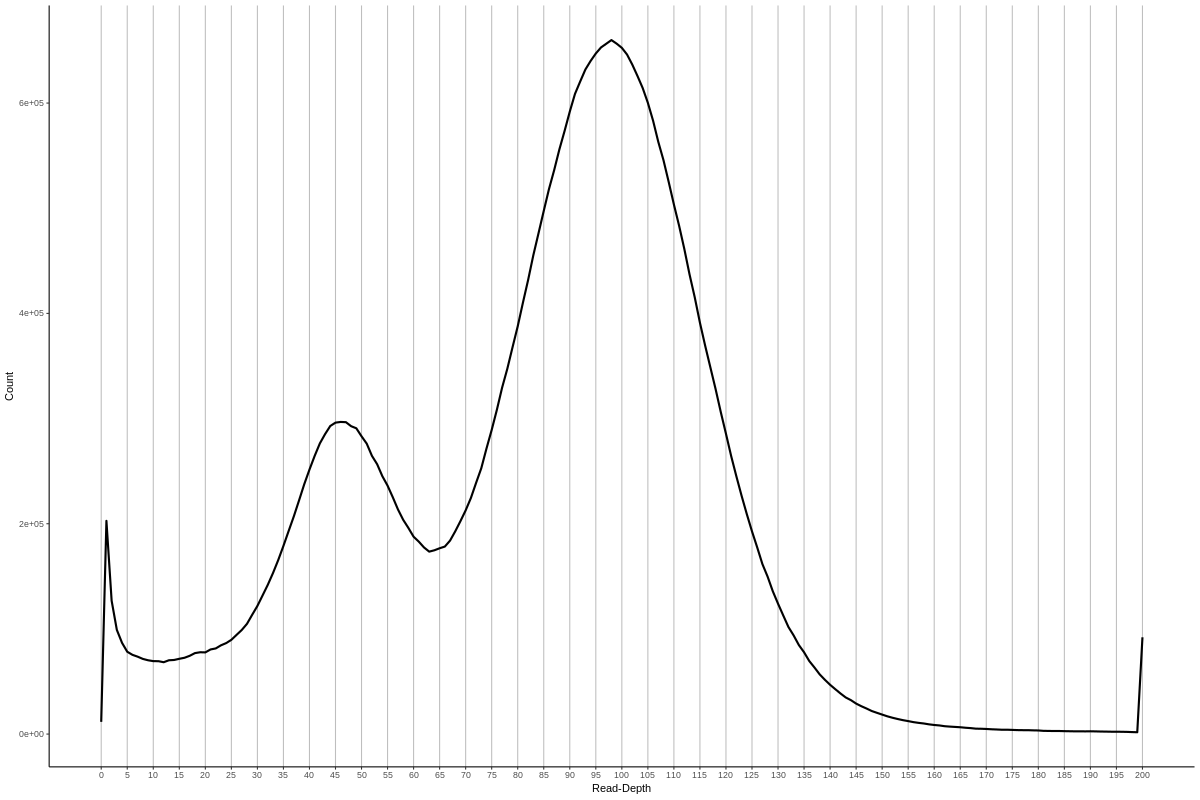
也就是下图,你能明显的看到图中有两个峰,一个是单倍型的覆盖度,另一个二倍型的覆盖度,
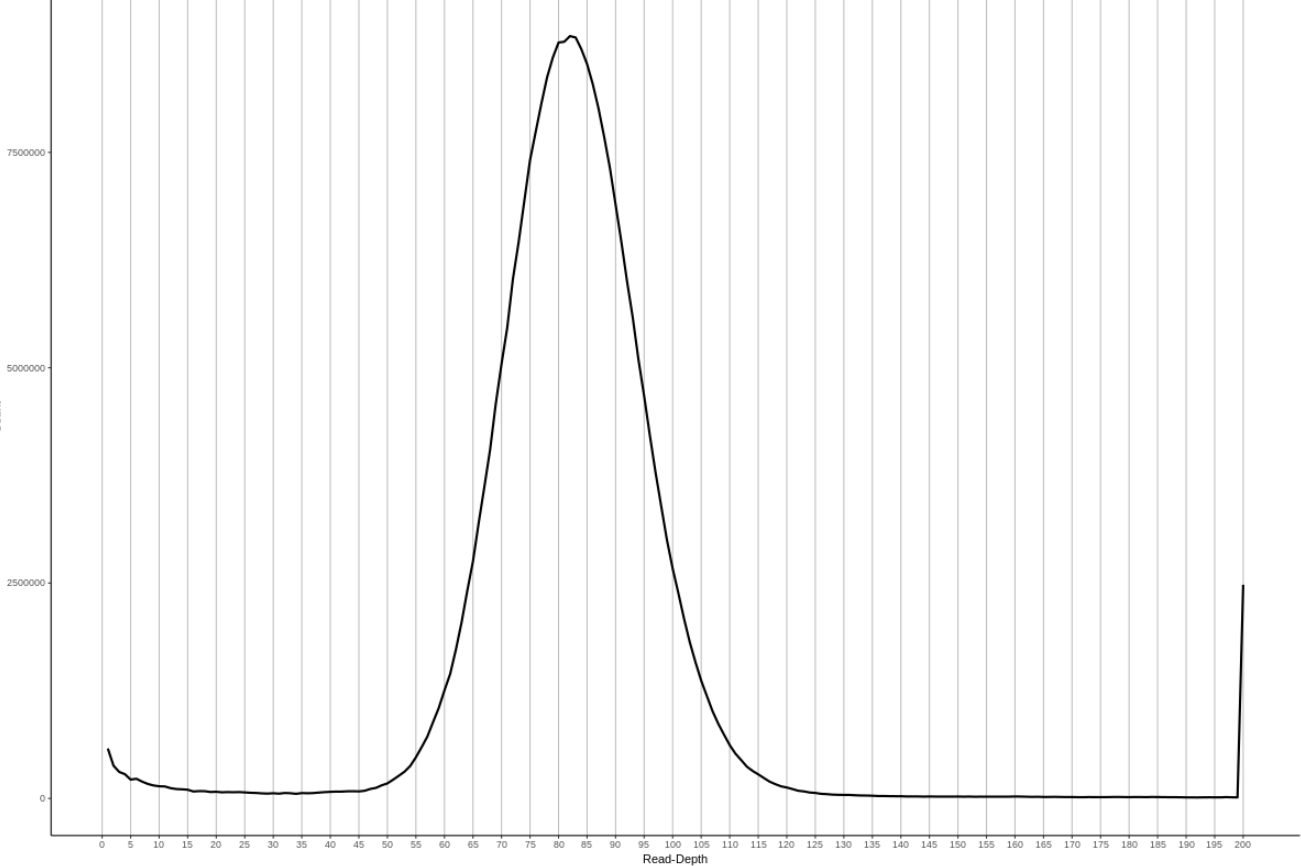
你可能还想知道高纯合基因组是什么样的效果,我也找了一个纯合的物种做了也做了read-depth 柱状图,
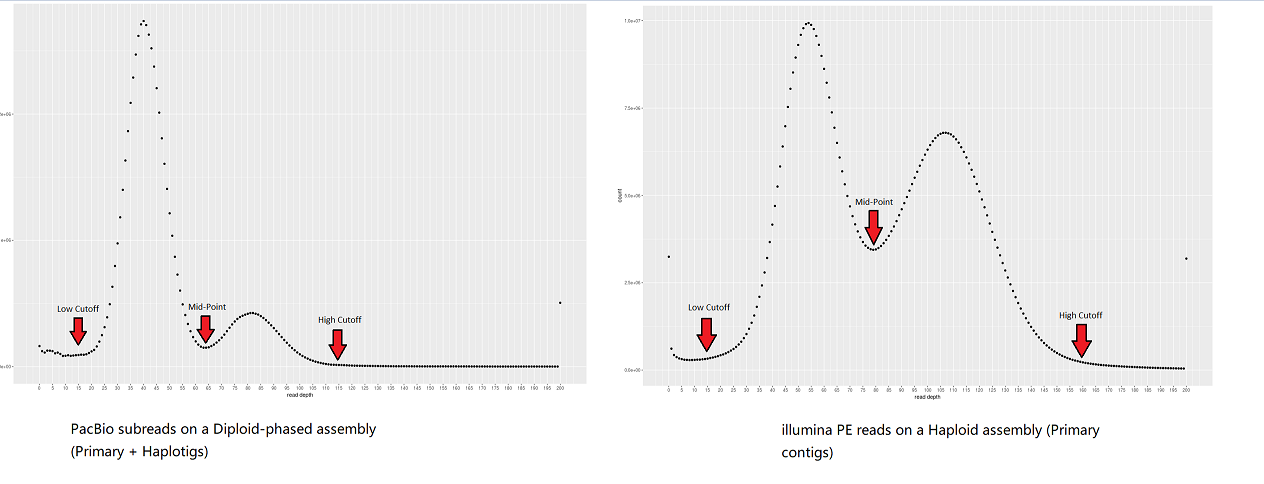
之后你需要根据read-depth 柱状图 确定这两个峰的位置用于下一步。下面是两个例子。对于我们则是,20,65,190.
第二步: 根据read-depth信息选择阈值。
purge_haplotigs contigcov -i cns_p_ctg.aligned.sd.bam.gencov -o coverage_stats.csv -l 20 -m 75 -h 190
这一步生成的文件是"coverage_stats.csv"
第三步:区分haplotigs.
purge_haplotigs purge -g cns_p_ctg.fasta -c coverage_stats.csv -b cns_p_ctg.aligned.sd.bam -t 4 -a 60
这一步会得到如下文件
- curated.artefacts.fasta:无用的contig,也就是没有足够覆盖度的contig.
- curated.fasta:新的单倍型组装
- curated.haplotigs.fasta:从原本组装分出来的haplotigs
- curated.reassignments.tsv: 单倍型的分配信息
- curated.contig_associations.log: 运行日志, 下面是其中一个记录,表示000004F_004和000004F_027是000004F_017的HAPLOTIG, 而000004F_017和000004F_013又是000004F,的HAPLOTIG。
000004F,PRIMARY -> 000004F_013,HAPLOTIG-> 000004F_017,HAPLOTIG -> 000004F_004,HAPLOTIG-> 000004F_027,HAPLOTIG
在"curated.reassignments.tsv"文件中有6列
- reassigned_contig: 用于比较的contig
- top_hit_contig: 最好的被比对的contig
- second_hit_contig: 第二个被比对的contig
- best_match_coverage: 最好的匹配覆盖度
- max_match_coverage : 最高的匹配深度
- reassignment: 标记为haplotype 还是 repeat,或者是keep
由于我们用的是单倍型组装primary contigs而不是二倍体组装的parimary + haplotigs, 因此我们需要将FALCON_Unzip的haplotgi合并到重新分配的haplotigs中,这样子我们依旧拥有二倍体组装结果
cat cns_h_ctg.fasta >> curated.haplotigs.fasta
原理介绍
为什么第一步的 read 覆盖深度分析能判断基因组是否冗余呢?这是因为对于坍缩的单倍型,那么含有等位的基因的read只能比对到该位置上,而如果杂合度太高被拆分成两个不同的contig,那么含有等位的基因的read就会分别比对到不同的read上,导致深度降低一半。下图A就是一个典型的包含冗余基因组的read覆盖度分布
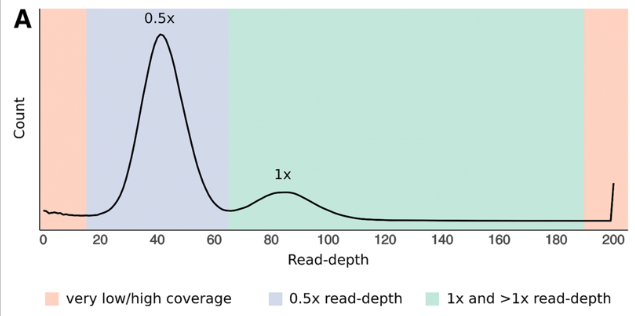
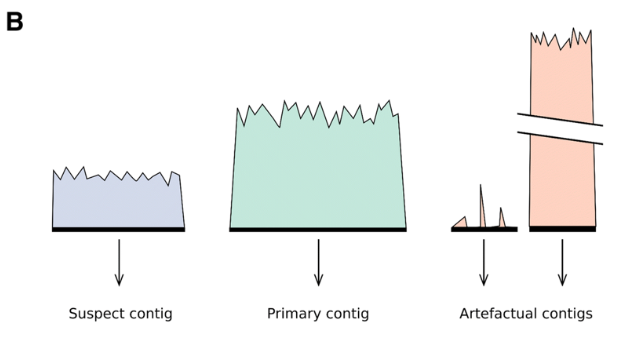
分析流程的第二步的任务就是人工划分出如下图B部分,绿色的部分是坍缩单倍型contig,蓝色的部分是潜在的冗余contig。之后,分析流程会计算这些区域中的contig的覆盖度。 对于绿色部分中的contig,如果覆盖度低于80%, 会进行标记用于后续分析。如果深度非常低,那么很有可能就是组装引入错误,深度非常高的部分基本就是重复序列或者是细胞器的contig,这些黄色的contig可以在后续的组装出分开。
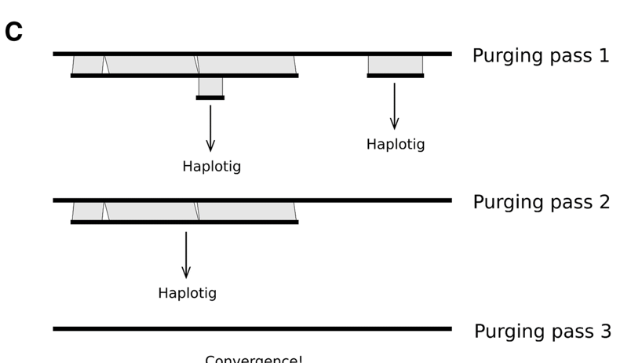
第三步就是同源序列进行识别和分配。所有标记的contig之后会用Minimap2在整个组装进行搜索,寻找相似度较高的离散区间(如下图C)。如果一个Contig的联配得分大于阈值(默认70%), 那么就会被标记为haplotigs. 如果一个contig的最大联配得分大于阈值(默认250%), 会被标记成重复序列,这有可能是潜在的有问题contig,或许是坍缩的contig或者低复杂度序列。
推荐阅读
- Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies
- https://bitbucket.org/mroachawri/purge_haplotigs
这篇关于使用purge_haplogs处理基因组杂合区域的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



