本文主要是介绍【单细胞转录组】使用Seurat的Read10X函数读取10x文件时为什么有格式要求,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
非模式物种在分析时可能会遇到各种各样的问题,但是也能学习到一些不太会注意到的小细节。比如我在分析数据的时候,使用seurat查找差异基因后绘图,这里展示一下某个cluster的结果:
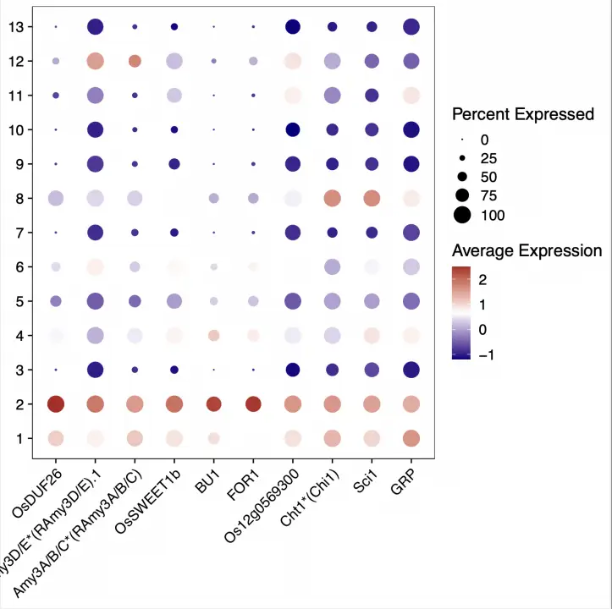
可以注意到出来Os号外还有一些看起来奇奇怪怪的基因,我本来以为是注释文件的问题。经师姐提醒,我去检查了一下features.tsv这个文件中的信息,发现这些长得奇奇怪怪的基因确实是存在的,而且是在第二列
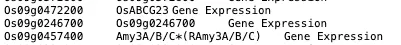
也可以在矩阵中检查。比如
matrix <- PRO@assays$RNA@data
tail(rownames(matrix),200)

说明在创建seurat对象的时候features信息自动读取的是第二列。
使用Read10X函数,可以直接读取三个文件,比如
pbmc.data <- Read10X(data.dir = "../data/pbmc3k/filtered_gene_bc_matrices/hg19/")
去看Read10X这个函数,同样会注意到gene.column这里默认是2,再次说明读取的是features.tsv这个文件的第二列。


这里要回顾一下cellranger(我目前是v7,提到版本是如果是很早的版本结果可能还不一样)输出的三个文件都是什么:
-
barcodes.tsv.gz
一列,细胞信息(列号)

-
features.tsv.gz
三列,基因信息(行号)
-
matrix.mtx.gz
三列,
第一列:行号
第二列:列号
第三列:表达量
代表某一行的基因在某一列的细胞中表达量是多少
了解到这些信息后,可以知道Read10X的功能,其实就是读取了一个矩阵,只是这个矩阵分成了三个部分:所有的细胞、所有的基因,以及它们的表达量信息。使用Read10X后这三个文件被还原成类似以下的样子:
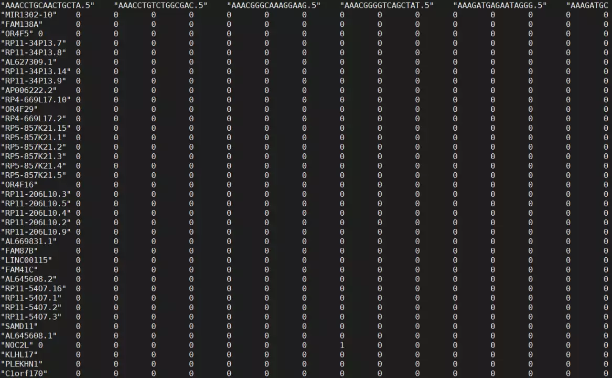
并且要注意,
- 基因默认是读取features.tsv.gz文件中的第二列(gene.column)
- 默认会过滤重复基因,只保留一个高丰度的(unique.features)
所以对于一些特殊物种,为了其它下游分析不出现bug(比如富集分析默认用的是geneid,但你在这里可能读的是symbol这一列),因此可以默认在读取矩阵的时候注意一下gene.column读的是哪一列,比如可以默认读第一列
reader_10x = Read10X(data.dir=dir, gene.column = 1)
这里再贴一下Read10X的源码
function (data.dir, gene.column = 2, cell.column = 1, unique.features = TRUE, strip.suffix = FALSE)
{full.data <- list()has_dt <- requireNamespace("data.table", quietly = TRUE) && requireNamespace("R.utils", quietly = TRUE)for (i in seq_along(along.with = data.dir)) {run <- data.dir[i]if (!dir.exists(paths = run)) {stop("Directory provided does not exist")}
#在data.dir路径下获得barcode、gene、features、matrix地址,并检查是否存在genes.tsv这个文件barcode.loc <- file.path(run, "barcodes.tsv")gene.loc <- file.path(run, "genes.tsv")features.loc <- file.path(run, "features.tsv.gz")matrix.loc <- file.path(run, "matrix.mtx")pre_ver_3 <- file.exists(gene.loc)if (!pre_ver_3) {addgz <- function(s) {return(paste0(s, ".gz"))}barcode.loc <- addgz(s = barcode.loc)matrix.loc <- addgz(s = matrix.loc)}if (!file.exists(barcode.loc)) {stop("Barcode file missing. Expecting ", basename(path = barcode.loc))}if (!pre_ver_3 && !file.exists(features.loc)) {stop("Gene name or features file missing. Expecting ", basename(path = features.loc))}if (!file.exists(matrix.loc)) {stop("Expression matrix file missing. Expecting ", basename(path = matrix.loc))}
#读取表达量data <- readMM(file = matrix.loc)if (has_dt) {
#读取细胞信息cell.barcodes <- as.data.frame(data.table::fread(barcode.loc, header = FALSE))}else {cell.barcodes <- read.table(file = barcode.loc, header = FALSE, sep = "\t", row.names = NULL)}if (ncol(x = cell.barcodes) > 1) {cell.names <- cell.barcodes[, cell.column]}else {cell.names <- readLines(con = barcode.loc)}if (all(grepl(pattern = "\\-1$", x = cell.names)) & strip.suffix) {
#对细胞信息进行修改,矩阵的colnames添加上细胞信息cell.names <- as.vector(x = as.character(x = sapply(X = cell.names, FUN = ExtractField, field = 1, delim = "-")))}if (is.null(x = names(x = data.dir))) {if (length(x = data.dir) < 2) {colnames(x = data) <- cell.names}else {colnames(x = data) <- paste0(i, "_", cell.names)}}else {colnames(x = data) <- paste0(names(x = data.dir)[i], "_", cell.names)}if (has_dt) {
#读取基因信息,并且命名genes.tsv/features.tsv.gz都是可以读取的feature.names <- as.data.frame(data.table::fread(ifelse(test = pre_ver_3, yes = gene.loc, no = features.loc), header = FALSE))}else {feature.names <- read.delim(file = ifelse(test = pre_ver_3, yes = gene.loc, no = features.loc), header = FALSE, stringsAsFactors = FALSE)}if (any(is.na(x = feature.names[, gene.column]))) {warning("Some features names are NA. Replacing NA names with ID from the opposite column requested", call. = FALSE, immediate. = TRUE)na.features <- which(x = is.na(x = feature.names[, gene.column]))replacement.column <- ifelse(test = gene.column == 2, yes = 1, no = 2)
#删除NA,并且选择读取第一列还是第二列feature.names[na.features, gene.column] <- feature.names[na.features, replacement.column]}if (unique.features) {fcols = ncol(x = feature.names)if (fcols < gene.column) {stop(paste0("gene.column was set to ", gene.column, " but feature.tsv.gz (or genes.tsv) only has ", fcols, " columns.", " Try setting the gene.column argument to a value <= to ", fcols, "."))}
#去重,矩阵的rownames添加上基因信息rownames(x = data) <- make.unique(names = feature.names[, gene.column])}if (ncol(x = feature.names) > 2) {data_types <- factor(x = feature.names$V3)lvls <- levels(x = data_types)if (length(x = lvls) > 1 && length(x = full.data) == 0) {message("10X data contains more than one type and is being returned as a list containing matrices of each type.")}expr_name <- "Gene Expression"if (expr_name %in% lvls) {lvls <- c(expr_name, lvls[-which(x = lvls == expr_name)])}data <- lapply(X = lvls, FUN = function(l) {return(data[data_types == l, , drop = FALSE])})names(x = data) <- lvls}else {data <- list(data)}full.data[[length(x = full.data) + 1]] <- data}list_of_data <- list()for (j in 1:length(x = full.data[[1]])) {list_of_data[[j]] <- do.call(cbind, lapply(X = full.data, FUN = `[[`, j))list_of_data[[j]] <- as.sparse(x = list_of_data[[j]])}names(x = list_of_data) <- names(x = full.data[[1]])if (length(x = list_of_data) == 1) {return(list_of_data[[1]])}else {return(list_of_data)}
}
这里还可以看到seurat为什么对命名有要求,如果未来遇到不是这样命名的,改这里就好。
barcode.loc <- file.path(run, "barcodes.tsv")gene.loc <- file.path(run, "genes.tsv")features.loc <- file.path(run, "features.tsv.gz")matrix.loc <- file.path(run, "matrix.mtx")
完~

本文由mdnice多平台发布
这篇关于【单细胞转录组】使用Seurat的Read10X函数读取10x文件时为什么有格式要求的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!




