本文主要是介绍单细胞分析(一)——seurat包单个样本处理,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
seurat包单个样本处理
- 10X genomics的基本原理
- 创建对象
- 加载数据
- 创建 Seurat 对象
- count matrix是什么样子?
- 预处理流程
- 计算线粒体含量
- 质控展示
- 正式筛选
- 数据标准化
- 特征筛选
- 寻找高变基因
- 高变基因绘图
- 缩放数据
- PCA线性降维
- 降维
- 降维展示
- UMAP非线性降维
- 聚类分群
- 参考文章
10X genomics的基本原理
大致如下
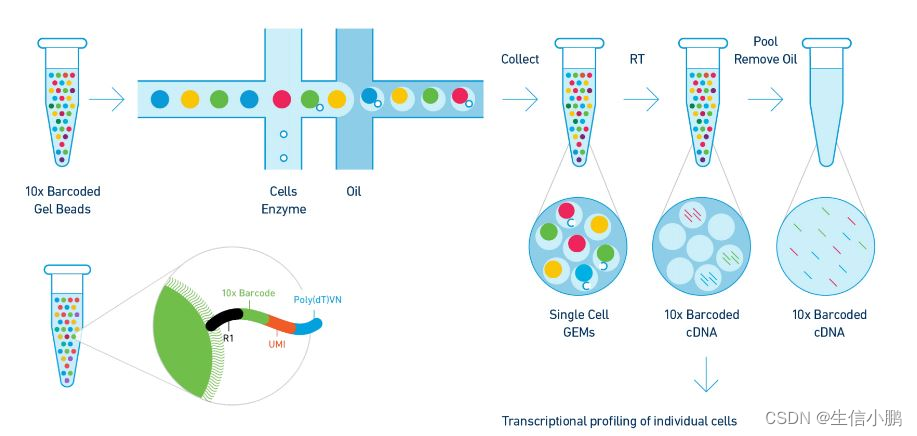 在这个教程中,主要将分析 10X Genomics 免费提供的外周血单核细胞 (PBMC) 数据集。在 Illumina NextSeq 500 上对 2,700 个单细胞进行了测序。可以在https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz此处找到原始数据。我们从读取数据开始。 Read10X() 函数从 10X 读取 cellranger 管道的输出,返回一个唯一的分子识别 (UMI) 计数矩阵。此矩阵中的值表示在每个单元格(列)中检测到的每个特征(即基因;行)的分子数。
在这个教程中,主要将分析 10X Genomics 免费提供的外周血单核细胞 (PBMC) 数据集。在 Illumina NextSeq 500 上对 2,700 个单细胞进行了测序。可以在https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz此处找到原始数据。我们从读取数据开始。 Read10X() 函数从 10X 读取 cellranger 管道的输出,返回一个唯一的分子识别 (UMI) 计数矩阵。此矩阵中的值表示在每个单元格(列)中检测到的每个特征(即基因;行)的分子数。
Read10X() 函数是针对于整理好的10X Genomics 数据,如果手头的不是类似文件,可以将其进行转换,成为格式一致的文件。
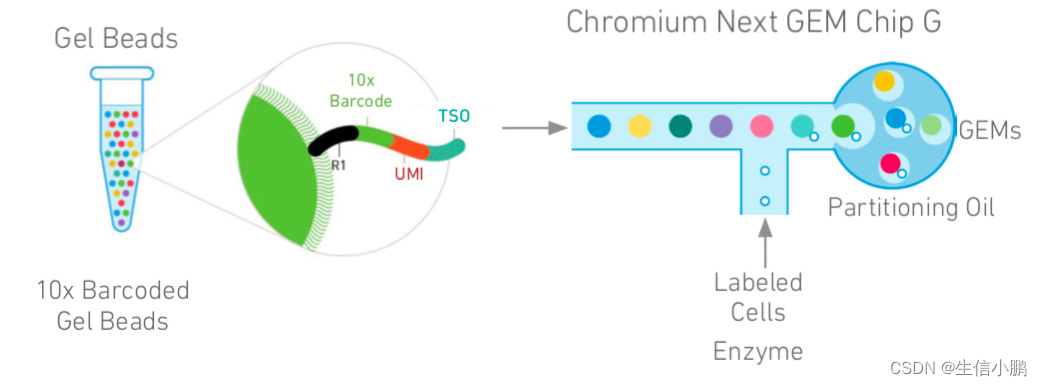
接下来使用计数矩阵创建一个 Seurat 对象。该对象用作包含单细胞数据集的数据(如计数矩阵)和分析(如 PCA 或聚类结果)的容器。例如,count matrix存储在 pbmc[[“RNA”]]@counts 中。
library(dplyr)
library(Seurat)
library(patchwork)
创建对象
加载数据
# Load the PBMC dataset
scobj <- Read10X(data.dir = "../data/pbmc3k/filtered_gene_bc_matrices/hg19/")
创建 Seurat 对象
### 2.创建Seurat对象
### counts 输入的是数据,行是基因,列是细胞
### project 参数输入的是项目名称,出现在metadata的orig.ident这一列
### min.cells 限定的是基因:每个基因在至少多少个细胞中出现
### min.features 限定的是细胞: 每个细胞中最少有多少个基因
scobj <- CreateSeuratObject(counts = scdata, project = "pbmc3k", min.cells = 3, min.features = 200)count matrix是什么样子?
count矩阵是稀松矩阵,可以减少占用空间
pbmc.data[c("IGF2BP2", "TCL1A", "MS4A1"), 1:30]dense.size <- object.size(as.matrix(pbmc.data))
dense.sizesparse.size <- object.size(pbmc.data)
sparse.sizedense.size/sparse.size
预处理流程
计算线粒体含量
这是质控的重要步骤,使用PercentageFeatureSet函数
### 主要PercentageFeatureSet函数计算线粒体含量
### 人类使用pattern = "^MT-",小鼠使用pattern = "^mt-"
scobj[["percent.mt"]] <- PercentageFeatureSet(scobj, pattern = "^MT-")### 该操作会在metadata数据里面增加一列叫做percent.mt
metadata <- scobj@meta.data一般情况下,可以认为线粒体含量多,意味着细胞可能趋于死亡,这样的细胞就应该剔除。但是如果本身研究的就是和线粒体相关的内容,例如药物干预会引起线粒体的变化,那么就要根据具体情况来分析是否需要剔除。
质控展示
### 质控数据可视化,使用VlnPlot函数
### nFeature_RNA, number of Feature, 每个细胞中有多少个基因
### nCount_RNA, number of counts, 每个细胞中有多少个counts
### percent.mt, 我们自己增加的列, 每个细胞中线粒体基因的比例
VlnPlot(scobj, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
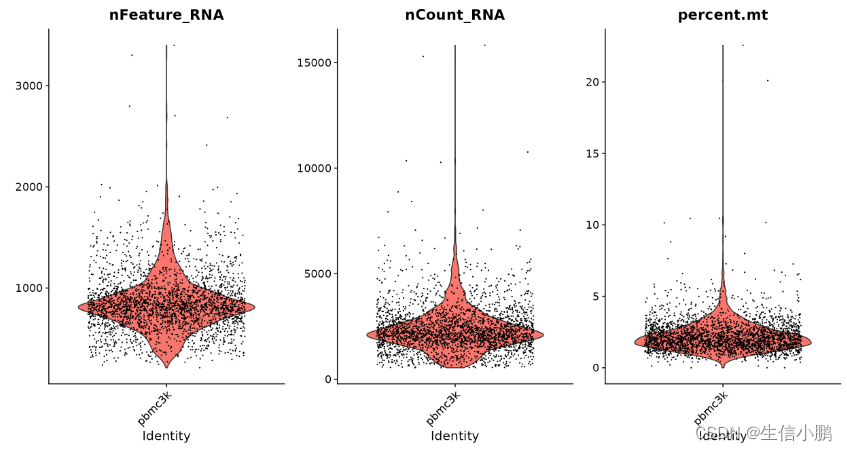
大部分情况下,线粒体的含量应该去除较大的,这个可以根据具体的情况进行分析,一般10%以内可以接受。但是依然是要根据相应的干预和特点进行取舍。
也可以展示特征之间的关系
# FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.plot1 <- FeatureScatter(scobj, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(scobj, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2

正式筛选
这一步依然是根据metadata的数据进行筛选,这一部分依然是根据相关的具体情况进行筛选。
### 正式筛选,筛选的是细胞,最终细胞减少
### nFeature_RNA > 200
### nFeature_RNA < 2500
### percent.mt < 5
scobj <- subset(scobj, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
筛选之后,再次用小提琴图展示,是上面小提琴图的缩减版本。
至于到底应该确定到多少的筛选指标,可以先按照常规指标进行设定,在后续的细胞聚类中,看看是否有与线粒体相关的群聚类出来,然后再更改筛选指标,聚类指标确实消失,那么就可以对筛选指标确定。
数据标准化
每个细胞分别进行检测,所以如果要比较各个细胞,还是要进行标准化
### 先除以总数,再乘以系数,然后取log
scobj <- NormalizeData(scobj, normalization.method = "LogNormalize", scale.factor = 10000)
### 默认参数可以省略
scobj <- NormalizeData(scobj)
特征筛选
如果某些基因的表达再各个细胞之间表达是恒定的,所以要区分各个细胞,就要使用变化差异大的基因。
寻找高变基因
scobj <- FindVariableFeatures(scobj, selection.method = "vst", nfeatures = 2000)
### 默认参数可以省略
scobj <- FindVariableFeatures(scobj)
高变基因绘图
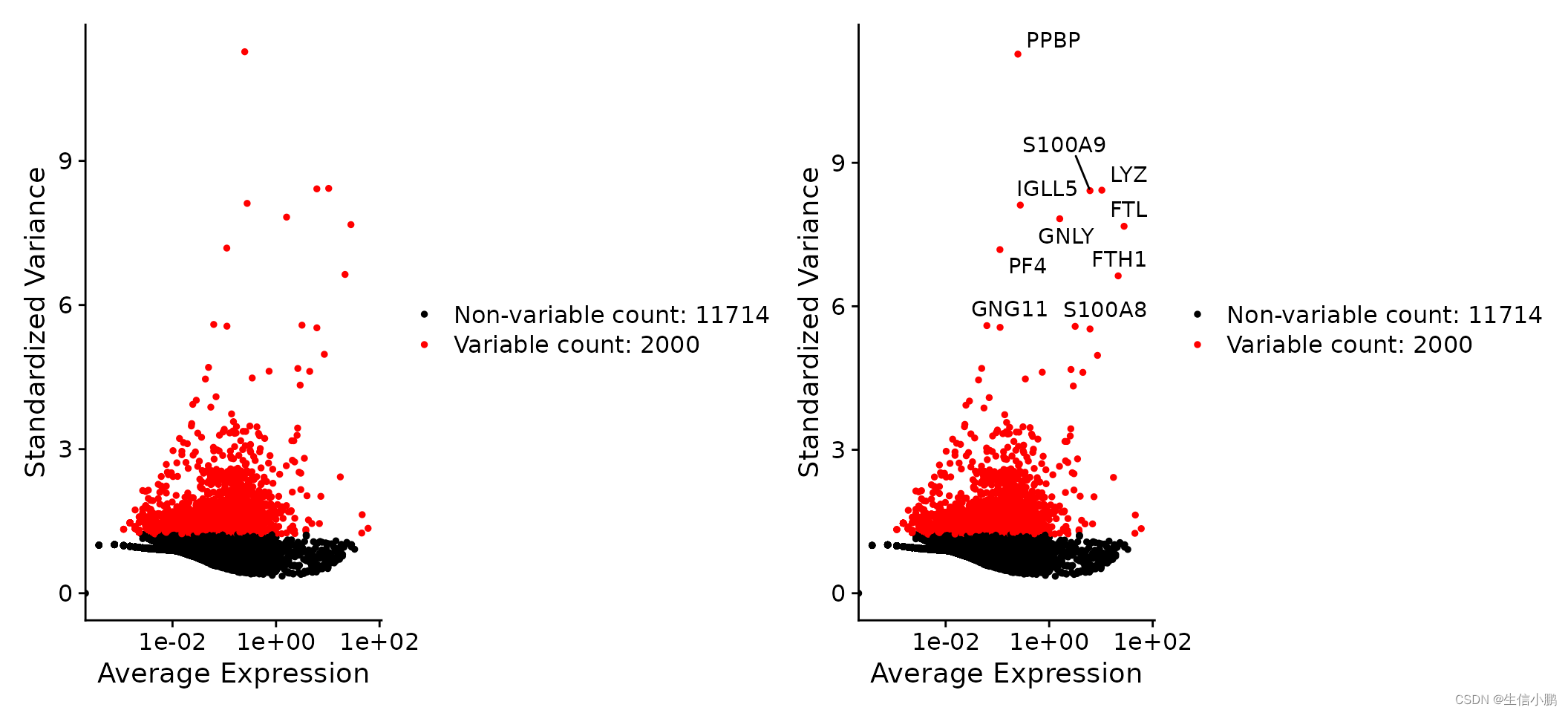
### 使用VariableFeatures 函数提取高变基因
### 等同于 scobj@assays$RNA@var.features
top10 <- head(VariableFeatures(scobj), 10)
### 使用VariableFeaturePlot 画图
plot1 <- VariableFeaturePlot(scobj)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
plot1 + plot2
缩放数据
缩放的数据达到的效果是:基因的平均值为0,方差为1。便于不同类型之间的比较。
### 降维之前的必备操作
scobj <- ScaleData(scobj, features = rownames(scobj))
### 如果不限定参数,只会缩放高变基因
### scobj <- ScaleData(scobj)
### 缩放后的数据存放在scobj@assays$RNA@scale.data,会很大
scale.data <- scobj@assays$RNA@scale.data
这一步缩放数据会花费比较长的时间,原因是这一步的操作是在所有基因上展开。但是,其实我们最需要的是变异大的基因,也就是可能是标志基因的数据。所以,这个函数默认是可以使用前2000的基因进行缩放的,可以省略features参数的内容。
scobj <- ScaleData(scobj)
需要说明的是,虽然缩放会花费一些时间,数据也会变大,但是,因为后续如果要绘制热图,使用的依然是缩放数据,所以依然在这一步将所有基因的数据进行缩放。
如果,保存数据的时候,还是可以把缩放数据去除后,再进行保存。下次需要的时候再次缩放即可
可以这样操作
scobj@assays$RNA@scale.data <-matrix()
PCA线性降维
PCA操作的对象为缩放过的数据,因此,需要保证前面的缩放数据。结果会保存在对象的reduction中,可以从中提取数据。
为什么要进行主成分分析,主要还是用少量的数据和维度,尽可能的保留原来数据特点。
降维
scobj <- RunPCA(scobj, features = VariableFeatures(object = scobj))
DimPlot(scobj, reduction = "pca")### PCA降维数据存放在scobj@reductions$pca中
pcadata = as.data.frame(scobj@reductions$pca@cell.embeddings)
ggplot(pcadata,aes(PC_1,PC_2,color="red"))+geom_point()### 选择合适的PCA维度
ElbowPlot(scobj)
降维展示
# Examine and visualize PCA results a few different ways
print(scobj[["pca"]], dims = 1:5, nfeatures = 5)
VizDimLoadings(scobj, dims = 1:2, reduction = "pca")

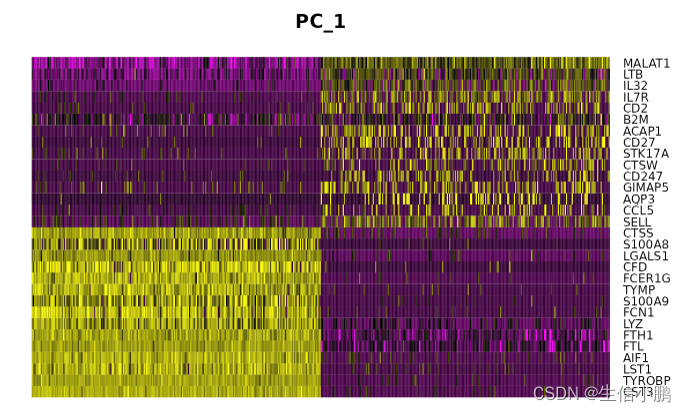
使用热图进行展示
DimHeatmap(scobj, dims = 1, cells = 500, balanced = TRUE)
dims 是确定需要展示维度数,可以尝试
UMAP非线性降维
如何将高维数据压缩到二维平面中,使用UMAP进行处理。其原理可以如下展示:
Understanding UMAP
### 依赖PCA的结果
### dims = 1:10 由上一步确定
scobj <- RunUMAP(scobj, dims = 1:10)
DimPlot(scobj, reduction = "umap")
### UMAP降维数据存放在scobj@reductions$umap中
umapdata = as.data.frame(scobj@reductions$umap@cell.embeddings)
ggplot(umapdata,aes(UMAP_1,UMAP_2,color="red"))+geom_point()
聚类分群
二维空间中看到了细胞分群,那么在这个基础上,怎么进行亚群分类,这就是这一步的意义。
### 找紧邻,dims = 1:10 跟UMAP相同
scobj <- FindNeighbors(scobj, dims = 1:10)
### 分群
scobj <- FindClusters(scobj, resolution = 0.5)
### 会在metadata中增加两列数据"RNA_snn_res.0.5" "seurat_clusters"
metadata <- scobj@meta.data
resolution是这个主要参数,也就是亚群切割的分辨率,这个具体怎么挑选,可以按照以下的方法可视化查看。
### 设置多个resolution选择合适的resolution
scobj <- FindClusters(scobj, resolution = seq(0.2,1.2,0.1))
metadata <- scobj@meta.data
library(clustree)
clustree(scobj)
根据上面的可视化结果,可以大致确定一个分辨率
### 选择特定分辨率得到的分群此处为RNA_snn_res.0.5
scobj@meta.data$seurat_clusters <- scobj@meta.data$RNA_snn_res.0.5
Idents(scobj) <- "seurat_clusters"
DimPlot(scobj, reduction = "umap", label = T)
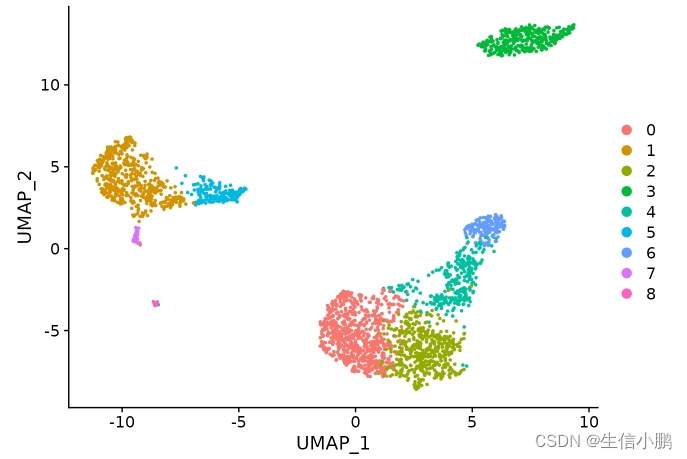
大致得到了相应的分群,后续就需要对这些分群进行注释。注释是一个与个人知识背景很相关的分析过程,后续补充。
参考文章
Seurat - Guided Clustering Tutorial
Visualization of gene expression with Nebulosa (in Seurat)
Use regularized negative binomial regression to normalize UMI count data
Tutorial: Integrating stimulated vs. control PBMC datasets to learn cell-type specific responses
Understanding UMAP
这篇关于单细胞分析(一)——seurat包单个样本处理的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!



