本文主要是介绍biostar handbook(五)|序列从何而来和质量控制,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
测序仪
2017年一篇发表在Nature的综述"DNA sequencing at 40: past, present and future"介绍了DNA测序这40年的发展历程。1976年,Sanger和Coulson同时发表了2种方法用于对上百个DNA碱基进行解码,这就是第一代测序技术。到了2005年,罗氏的454平台揭开了高通量测序的序幕,后面则是SOLiD,454和Illumina三方对抗。然而10年过去了,市面上的二代测序只有illumina一家独大。在三代测序技术或者4代测序技术上,目前则是PacBio和MinION占领了市场的大部分份额。
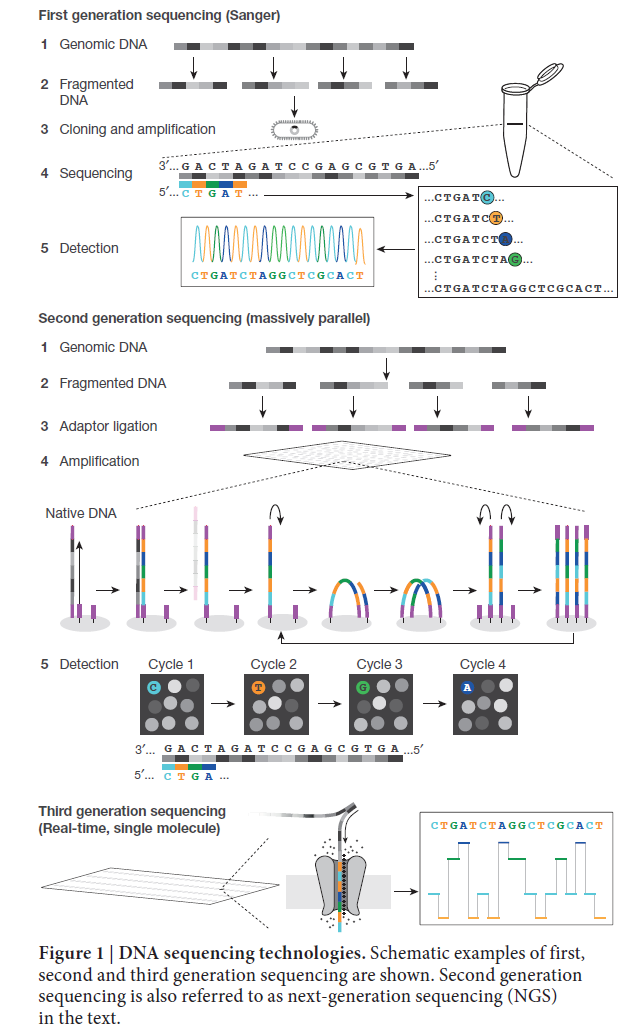
测序仪的工作原理
高通量测序之所以能够能够达到如此高的通量的原因就是他把原来几十M,几百M,甚至几个G的基因组通过物理或化学的方式打算成几百bp的短序列,然后同时测序。
因为测序过程是边合成边测序(SBS),所以在建库的时候,短序列两段会加一些固定的碱基用于桥式PCR扩增,这些固定的碱基就是adapter(接头)。一般而言,还可以在接头加一些tag(index),用于标识这个read来自于哪个物种。目前的单细胞测序为了省钱,譬如10X genomic技术,都是在一个pool里面加多种接头。
以二代测序的无冕之王illumina测序仪建库为例,假设有如下的DNA片段
AAAATTTTGGGGCCCC
TTTTAAAACCCCGGGG
在建库准备时,单一设计的DNA接头长度通常超过30个碱基,会加在每条序列的两端
XXXXAAAATTTTGGGGCCCCYYYY
XXXXGGGGCCCCAAAATTTTYYYY
对于给定的片段,每条链都会被测序。测序仪通常会识别起始位点的XXXX,但不会被记录,测序方向:
---->
AAAAT
AAAATTTTGGGGCCCC
TTTTAAAACCCCGGGGCGGGG<----
由于片段长度不一,所以会出现read-through的情况,也就是读到了接头的序列。
由于测序的protocol和instrumentation不同,测序方向也可能不同,这会导致程序出现错误。因此请确保你的双端数据的方向是----> <----。
在测序过程中,机器会对每次读取的结果赋予一个值,用于表明它有多大把握结果是对的。从理论上都是前面质量好,后面质量差。并且在某些GC比例高的区域,测序质量会大幅度降低。
目前,Illumina的错误率是1/1000,PacBio是1/10,而MinION是1/5。随着技术的更新,目前它们的错误率应该是得到很大的降低了,但是顺序不会变,于是三代和四代的测序结果一般都需要二代进行纠错。
测序仪详解
2011年有一篇文章Travis Glenn’s Field Guide to Next Generation DNA Sequencer对不同测序仪进行了测评。现在6年过去了,这篇文章的内容也得到了相应的更新,见2016: Updates to the NGS Field Guide
Ilumina
待补充
PacBio
待补充
Minion
待补充
样本准备
待补充
测序数据覆盖度
什么叫做测序数据的覆盖度(coverage),这是一个很好的问题。在书中,覆盖率简单定义为:
c = 测序的碱基数 / 基因组总大小
一开始我觉得这个公式其实是计算测序的平均深度。但是后面继续谈到覆盖度不是意味着所有基因组都被覆盖了,而是覆盖率越高,基因组未被检测到的基因越少。根据经验公式,碱基丢失率:P = exp(-C)。假设测序深度10x,基因组长度为20k,那么丢失exp(-10)*20000,差不多是一个碱基,如果是人类基因组会是136199个碱基。
当然理论覆盖度并不代表现实情况,由于基因组的复杂性,DNA可能也不是真的随机打断,甚至实验protocol还有一定的偏向性。
- 尽可能增加测序深度
- 尽管有一些基因组部分很难被测序,但是我们其实清楚这些区域难以测序的原因
- 基因组的高度重复区域需要更长的读长才能被发现
- 基因组不同区域可能会产生相同的read,你需要更长的读长。
科学家喜欢用“可进入(accessible)", "可比对(mappable)", "有效(effective)"的基因组来指明基因组哪些区域很容易被研究。
数据质量和质量控制
一般而言,拿到数据后最重要的一步就是看看数据的质量如何。之前已经提及过FASTQ的基本格式,这里就不具体展开,并且我们其实一般都是使用Babraham Institute开发的FastQC对质量进行可视化展示。
目前来看,FastQC基本上已经是数据质量展示的通用工具了。它使用Java进行开发,是跨平台工具,效率高,简单易用,出图也好看。但是记住一点,FastQC不做质量控制,它只是展示数据的质量而已。
使用FastQC展示数据质量
FastQC的工作原理是通过对总体数据的抽样来评估总体效果,这就是它快(fast)的愿意,毕竟其他一些质量展示软件是老老实实把所有数据都用于作图。
首先你需要准备数据和软件:
conda -c bioconda install fastqc
wget http://data.biostarhandbook.com/data/sequencing-platform-data.tar.gz
tar xzvf sequencing-platform-data.tar.gz
然后你需要稍微了解以下fastqc的参数:
fastqc seqfile1 seqfile2 .. seqfileN
常用参数:
-o: 输出路径
--extract: 输出文件是否需要自动解压 默认是--noextract
-t: 线程, 和电脑配置有关,每个线程需要250MB的内存
-c: 测序中可能会有污染, 比如说混入其他物种
-a: 接头
-q: 安静模式
最后使用FastQC进行数据质量可视化展示
fastqc *.fq
结果会得到每个FASTQ文件对应的zip压缩文件和HTML文件。数据汇总主要会用ZIP压缩文件,而对数据质量的直观感受则是看HTML文件,直接用网页打开。绿色表示通过,红色表示未通过,黄色表示不太好
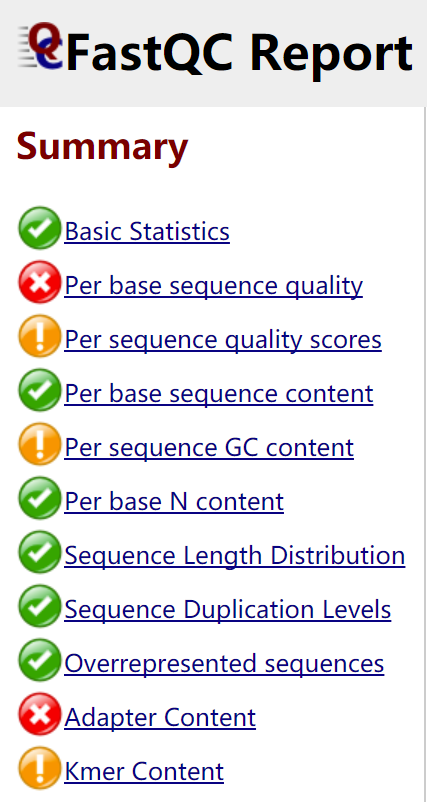
具体含义可以看这里: http://jingyan.baidu.com/article/49711c6149e27dfa441b7c34.html
一些注意事项:
- 没必要太过在意“stoplinght",但是如果全部红灯,那么数据就要小心了。
- 序列重复分为两类:天然重复(片段相同),人为重复(PCR扩增,检测)
检测重复有两种方法:序列相同,比对相同。读段重复最大的问题是在检测变异上,如果一个变异点重复两次,会产生与实际不符的效果。SNP calling和基因组变异检测需要移除重复,其他就不需要。
如果同一批测序有多个数据,比如说15个(5个样本,3个重复),在对每个数据做一个fastqc后,还可以用multiqc进行数据聚合展示
利用conda安装软件尤其简单,
conda install multiqc
multiqc --help
使用也很方便,
# 先获取QC结果ls *gz | while read id; do fastqc -t 4 $id; done
# multiqc
multiqc *fastqc.zip --pdf
会有一个html文件用来了解总体情况
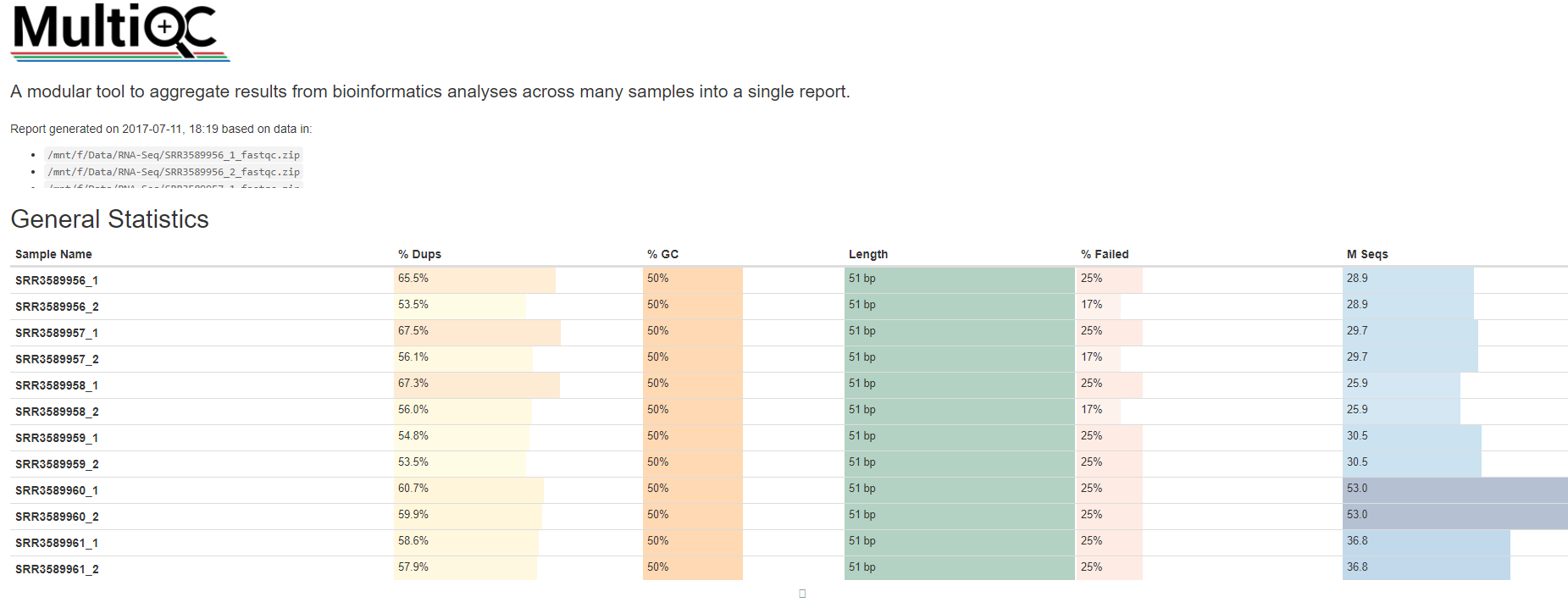
测序质量的质量控制
质控时机:比对前的原始数据和比对后的数据过滤
流程:
- 数据可视化评估
- 质量不错就停止QC
- 否则对数据进行修改,返回步骤1
QC工具的可信度
- 首先QC工具本身质量就不是很好,QC工具之间可能也不一致,不同工具使用相同的参数可能也会有不同的结果。
- QC的确可能会引入错误,所以如果尽量避免修改数据
QC工具集
比较好的是Trimmomatic, BBDuk ,flexbar and cutadapt
- BBDuk part of the BBMap package
- BioPieces a suite of programs for sequence preprocessing
- CutAdapt application note in Embnet Journal, 2011
- fastq-mcf published in The Open Bioinformatics Journal, 2013
- Fastx Toolkit: collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing - one of the first tools
- FlexBar, Flexible barcode and adapter removal published in Biology, 2012
- NGS Toolkit published in Plos One, 2012
- PrinSeq application note in Bioinformatics, 2011
- Scythe a bayesian adaptor trimmer
- SeqPrep - a tool for stripping adaptors and/or merging paired reads with overlap into single reads.
- Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads.
- TagCleaner published in BMC Bioinformatics, 2010
- TagDust published in Bioinformatics, 2009
- Trim Galore - a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files, with some extra functionality for MspI-digested RRBS-type (Reduced Representation Bisufite-Seq) libraries
- Trimmomatic application note in Nucleic Acid Research, 2012, web server issue
There also exist libraries via R (Bioconductor) for QC: PIQA, ShortRead
这篇关于biostar handbook(五)|序列从何而来和质量控制的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







