本文主要是介绍「JCVI教程」如何基于物种的CDS的blast结果绘制点图(dotplot),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
这是唐海宝老师GitHub上的JCVI工具的非官方说明书。
该工具集的功能非常多,但是教程资料目前看起来并不多,因此为了能让更多人用上那么好用的工具,我就一边探索,一边写教程
这一篇文章教大家如何利用JCVI里面的工具绘制点图,展现两个物种之间的共线性关系。
在分析之前,你需要从PhytozomeV11 下载A.thaliana和Alyrata的CDS序列,保证文件夹里有如下内容
Alyrata_384_v2.1.cds.fa.gz Athaliana_167_TAIR10.cds.fa.gz
Alyrata_384_v2.1.gene.gff3.gz Athaliana_167_TAIR10.gene.gff3.gz
准备最长CDS和BED文件
我们在做CDS相互比对的时候只需要有每个基因最长的转录本即可,有两种方法可以实现
方法1:自己写脚本
我用我写的一个脚本get_the_longest_transcripts.py提取每个基因的最长转录本,见 基因组共线性工具MCScanX使用说明
zcat Alyrata_384_v2.1.gene.gff3.gz | python ~/scripts/python/get_the_longest_transcripts.py > aly_lst_gene.txt
zcat Athaliana_167_TAIR10.gene.gff3.gz | python ~/scripts/python/get_the_longest_transcripts.py > ath_lst_gene.txt
其中xxx_lst_gene.txt的格式如下, 第一列是基因名,第二列是mRNA编号,后面几列是位置信息。
$ head ath_lst_gene.txt
AT4G19470.TAIR10 AT4G19470.1.TAIR10 Chr4 10612993 10614339 -
AT5G43860.TAIR10 AT5G43860.1.TAIR10 Chr5 17630450 17632312 +
AT1G68650.TAIR10 AT1G68650.1.TAIR10 Chr1 25775741 25777874 +
AT1G28050.TAIR10 AT1G28050.1.TAIR10 Chr1 9775528 9777810 -
AT3G59880.TAIR10 AT3G59880.1.TAIR10 Chr3 22120969 22121700 +
AT1G22030.TAIR10 AT1G22030.1.TAIR10 Chr1 7759164 7760556 -
AT5G24330.TAIR10 AT5G24330.1.TAIR10 Chr5 8295147 8297068 -
AT5G43990.TAIR10 AT5G43990.2.TAIR10 Chr5 17697889 17702005 +
AT1G11410.TAIR10 AT1G11410.1.TAIR10 Chr1 3841286 3844432 +
AT4G32890.TAIR10 AT4G32890.1.TAIR10 Chr4 15875470 15876762 +
由于基因名和mRNA编号里有在提取CDS不需要的内容,因此要进行删除
sed -i 's/\.v2\.1//g' aly_lst_gene.txt
sed -i 's/\.TAIR10//g' ath_lst_gene.txt
之后我们就可以根据第二列进行提取CDS
seqkit grep -f <(cut -f 2 ath_lst_gene.txt ) Athaliana_167_TAIR10.cds.fa.gz > ath.cds
seqkit grep -f <(cut -f 2 aly_lst_gene.txt ) Alyrata_384_v2.1.cds.fa.gz > aly.cds
提取的CDS编号里面也有一些不需要的内容,所以也要删除
sed -i 's/\.t.*//' aly.cds
sed -i 's/\..*//' ath.cds
此外还需要基因的位置信息的bed文件
awk '{print $3"\t"$4"\t"$5"\t"$1"\t0\t"$6}' ath_lst_gene.txt | sort -k4,4V > ath.bed
awk '{print $3"\t"$4"\t"$5"\t"$1"\t0\t"$6}' aly_lst_gene.txt | sort -k4,4V > aly.bed
基于JCVI工具集
当然也可以参考「JCVI教程」如何基于编码序列或蛋白序列进行共线性分析来提取bed和cds序列,不需要用到我写的脚本。
python -m jcvi.formats.gff bed --type=mRNA --key=Name Athaliana_167_TAIR10.gene.gff3.gz > ath.bed
python -m jcvi.formats.gff bed --type=mRNA --key=Name Alyrata_384_v2.1.gene.gff3.gz > aly.bed
对bed文件中的基因进行去重
python -m jcvi.formats.bed uniq ath.bed
python -m jcvi.formats.bed uniq aly.bed
这一步会得到aly.uniq.bed和ath.uniq.bed, 我们将其覆盖原文件
mv ath.uniq.bed ath.bed
mv aly.uniq.bed aly.bed
根据bed文件提取cds里的序列
# Athaliana
seqkit grep -f <(cut -f 4 ath.bed ) Athaliana_167_TAIR10.cds.fa.gz | seqkit seq -i > ath.cds
# Alyrata
seqkit grep -f <(cut -f 4 aly.bed ) Alyrata_384_v2.1.cds.fa.gz | seqkit seq -i > aly.cds
无论是哪种方法,请保证最后有以下四个文件
$ ls ???.???
aly.bed aly.cds ath.bed ath.cds
BLAST比对
相对于上一步,这一步其实非常简单了
makeblastdb -in ath.cds -out db/ath -dbtype nucl
blastn -num_threads 20 -query aly.cds -db db/ath -outfmt 6 -evalue 1e-5 -num_alignments 5 > aly_ath.blast
用jcvi.compara.blastfilter对结果进行过滤
python -m jcvi.compara.blastfilter --no_strip_names aly_ath.blast --sbed ath.bed --qbed aly.bed
运行过程中有如下输出信息
19:59:15 [base] Load file `aly.bed`
19:59:16 [base] Load file `ath.bed`
19:59:16 [blastfilter] Load BLAST file `aly_ath.blast` (total 49887 lines)
19:59:16 [base] Load file `aly_ath.blast`
19:59:16 [blastfilter] running the cscore filter (cscore>=0.70) ..
19:59:16 [blastfilter] after filter (42023->26531) ..
19:59:16 [blastfilter] running the local dups filter (tandem_Nmax=10) ..
19:59:16 [blastfilter] after filter (26531->24242) ..
最后输出aly_ath.blast.filtered用于做图
python -m jcvi.graphics.blastplot aly_ath.blast.filtered --sbed ath.bed --qbed aly.bed
最后点图如下
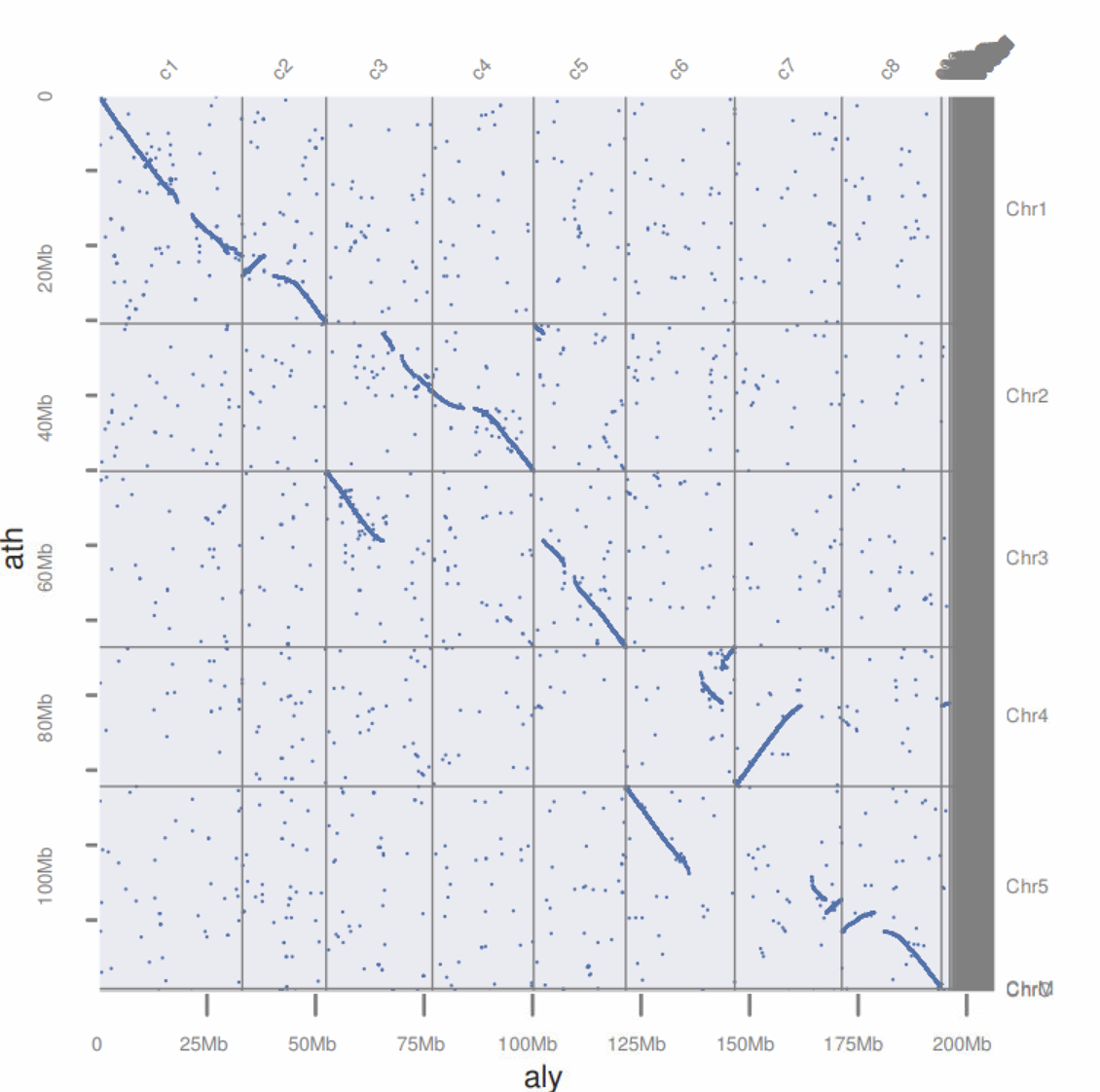
这篇关于「JCVI教程」如何基于物种的CDS的blast结果绘制点图(dotplot)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!









