本文主要是介绍单细胞|RNA-seq ATAC-seq 联合分析,希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
引言
本文[1]将介绍如何利用Signac和Seurat这两个工具,对一个同时记录了DNA可接触性和基因表达的单细胞数据集进行综合分析。我们将以一个公开的10x Genomics Multiome数据集为例,该数据集针对的是人体的外周血单核细胞。
数据准备
library(Signac)
library(Seurat)
library(EnsDb.Hsapiens.v86)
library(BSgenome.Hsapiens.UCSC.hg38)
# load the RNA and ATAC data
counts <- Read10X_h5("../vignette_data/multiomic/pbmc_granulocyte_sorted_10k_filtered_feature_bc_matrix.h5")
fragpath <- "../vignette_data/multiomic/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz"
# get gene annotations for hg38
annotation <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevels(annotation) <- paste0('chr', seqlevels(annotation))
# create a Seurat object containing the RNA adata
pbmc <- CreateSeuratObject(
counts = counts$`Gene Expression`,
assay = "RNA"
)
# create ATAC assay and add it to the object
pbmc[["ATAC"]] <- CreateChromatinAssay(
counts = counts$Peaks,
sep = c(":", "-"),
fragments = fragpath,
annotation = annotation
)
pbmc
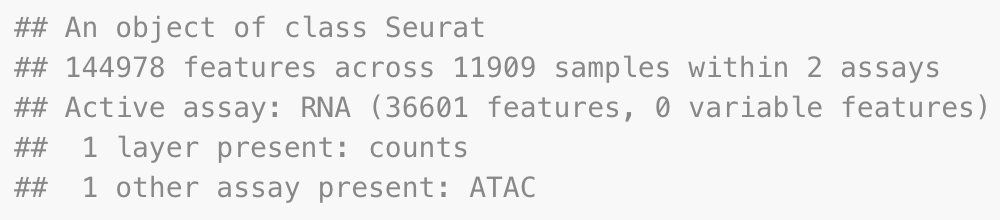
质控
我们可以通过DNA可及性数据来评估每个细胞的质量控制指标,并排除那些指标异常的细胞。此外,对于那些在RNA或ATAC检测中计数特别低或特别高的细胞,我们也会进行剔除。
DefaultAssay(pbmc) <- "ATAC"
pbmc <- NucleosomeSignal(pbmc)
pbmc <- TSSEnrichment(pbmc)
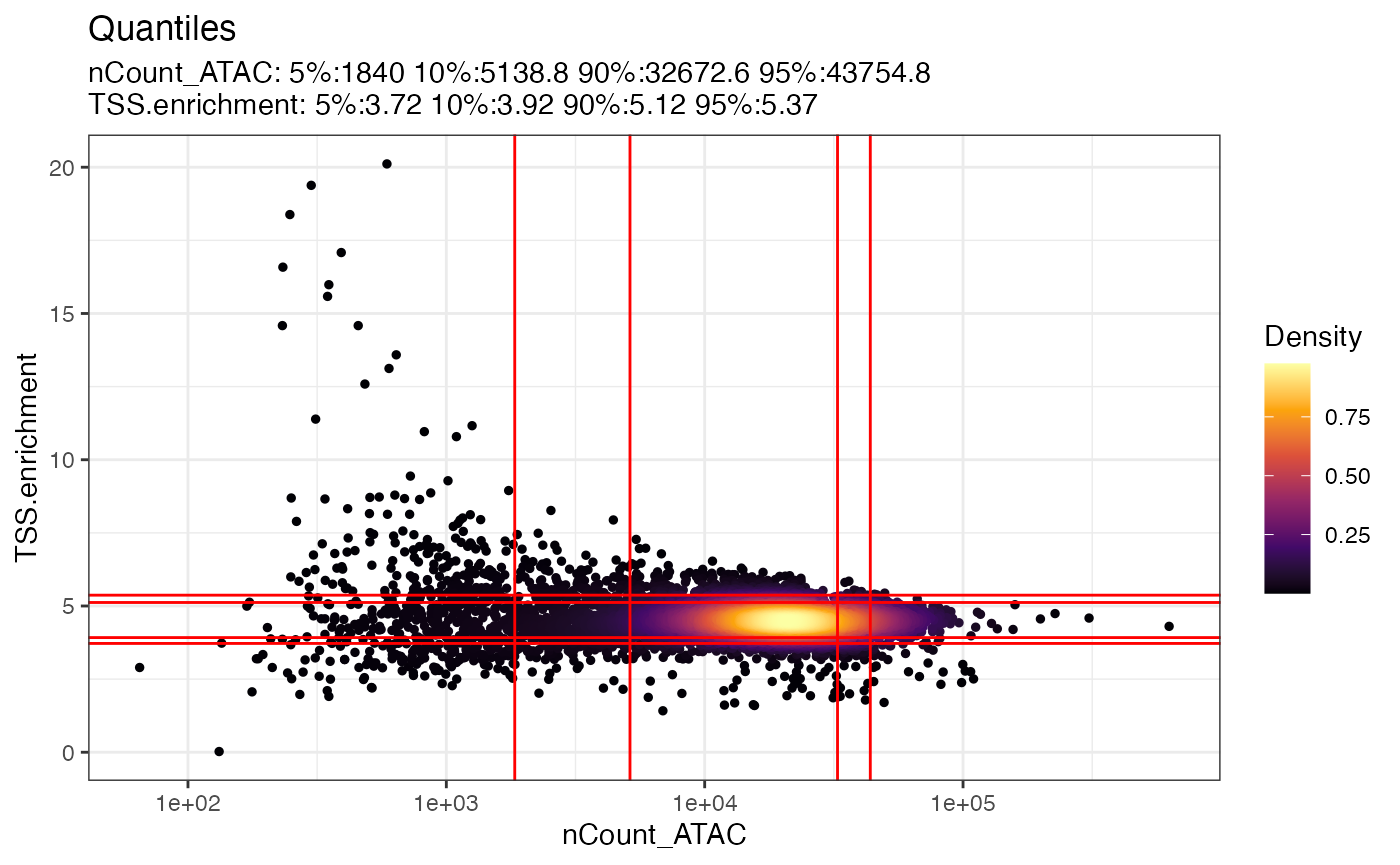
对象数据中变量之间的相互关系可以通过 DensityScatter() 函数来直观展示。此外,设置 quantiles=TRUE 选项,可以帮助我们迅速确定不同质量控制指标的适宜阈值。
DensityScatter(pbmc, x = 'nCount_ATAC', y = 'TSS.enrichment', log_x = TRUE, quantiles = TRUE)
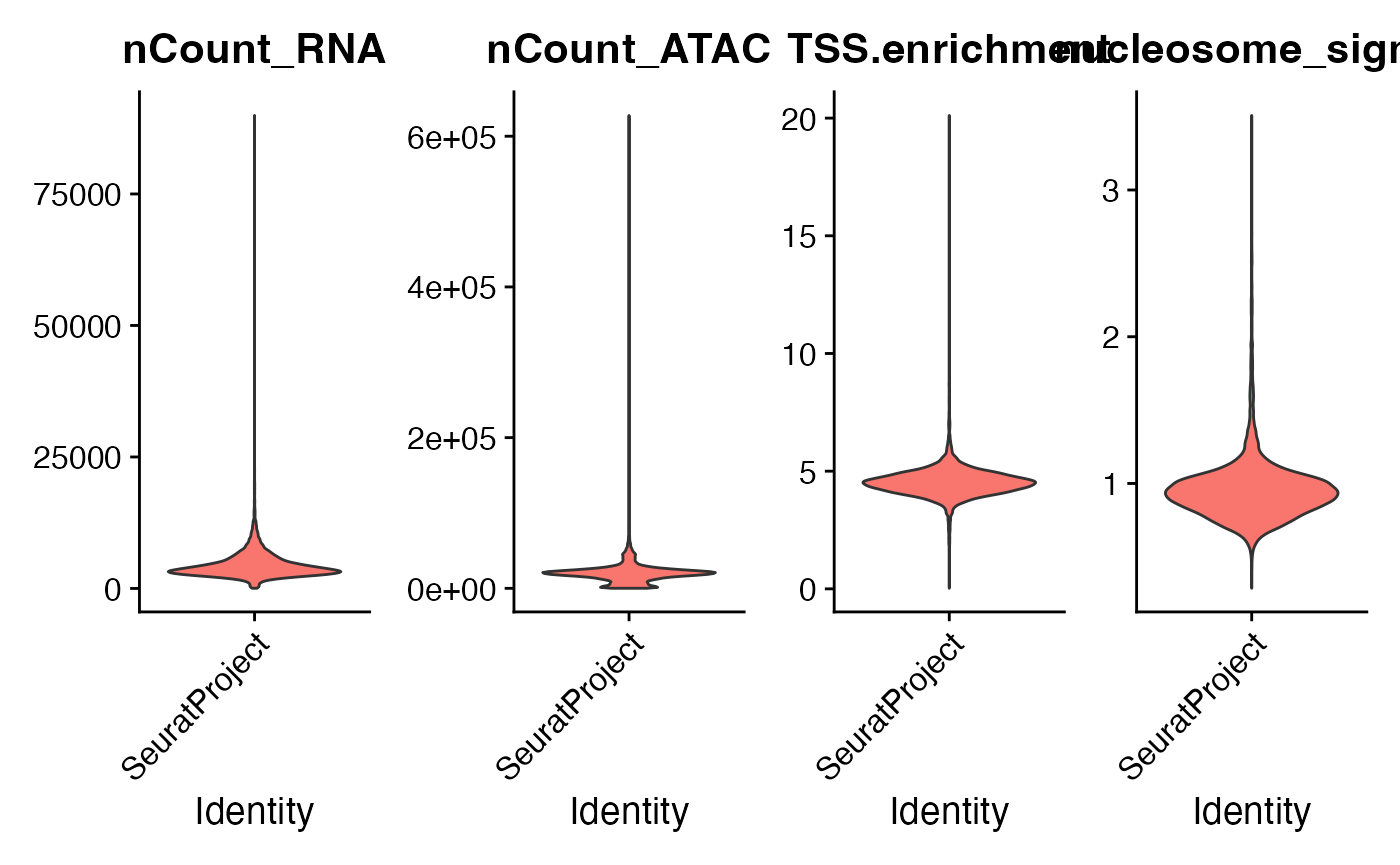
VlnPlot(
object = pbmc,
features = c("nCount_RNA", "nCount_ATAC", "TSS.enrichment", "nucleosome_signal"),
ncol = 4,
pt.size = 0
)
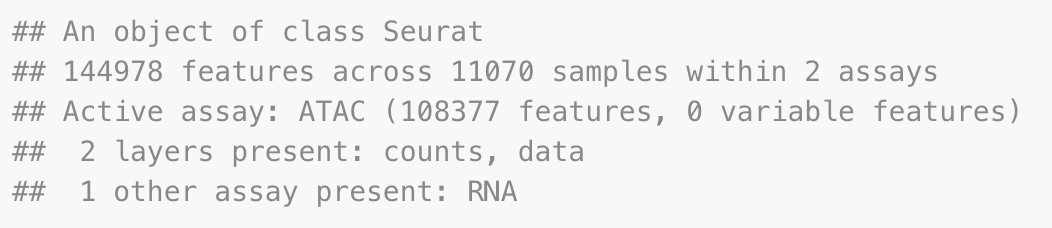
# filter out low quality cells
pbmc <- subset(
x = pbmc,
subset = nCount_ATAC < 100000 &
nCount_RNA < 25000 &
nCount_ATAC > 1800 &
nCount_RNA > 1000 &
nucleosome_signal < 2 &
TSS.enrichment > 1
)
pbmc
基因表达数据处理
我们可以使用 SCTransform 对基因表达数据进行标准化,并使用 PCA 降低维度。
DefaultAssay(pbmc) <- "RNA"
pbmc <- SCTransform(pbmc)
pbmc <- RunPCA(pbmc)
DNA可及性数据处理
在这里,我们通过执行潜在语义索引 ( LSI ),以处理 scATAC-seq 数据集的相同方式处理 DNA 可及性检测。
DefaultAssay(pbmc) <- "ATAC"
pbmc <- FindTopFeatures(pbmc, min.cutoff = 5)
pbmc <- RunTFIDF(pbmc)
pbmc <- RunSVD(pbmc)
注释细胞类型
为了注释数据集中的细胞类型,我们可以使用 Seurat 包中的工具,将细胞标签从现有的 PBMC 参考数据集中转移过来。
我们将使用 Hao 等人(2020年)的注释 PBMC 参考数据集,可以从这里下载:https://atlas.fredhutch.org/data/nygc/multimodal/pbmc_multimodal.h5seurat
请注意,加载参考数据集需要安装 SeuratDisk 包。
library(SeuratDisk)
# load PBMC reference
reference <- LoadH5Seurat("../vignette_data/multiomic/pbmc_multimodal.h5seurat", assays = list("SCT" = "counts"), reductions = 'spca')
reference <- UpdateSeuratObject(reference)
DefaultAssay(pbmc) <- "SCT"
# transfer cell type labels from reference to query
transfer_anchors <- FindTransferAnchors(
reference = reference,
query = pbmc,
normalization.method = "SCT",
reference.reduction = "spca",
recompute.residuals = FALSE,
dims = 1:50
)
predictions <- TransferData(
anchorset = transfer_anchors,
refdata = reference$celltype.l2,
weight.reduction = pbmc[['pca']],
dims = 1:50
)
pbmc <- AddMetaData(
object = pbmc,
metadata = predictions
)
# set the cell identities to the cell type predictions
Idents(pbmc) <- "predicted.id"
# remove low-quality predictions
pbmc <- pbmc[, pbmc$prediction.score.max > 0.5]
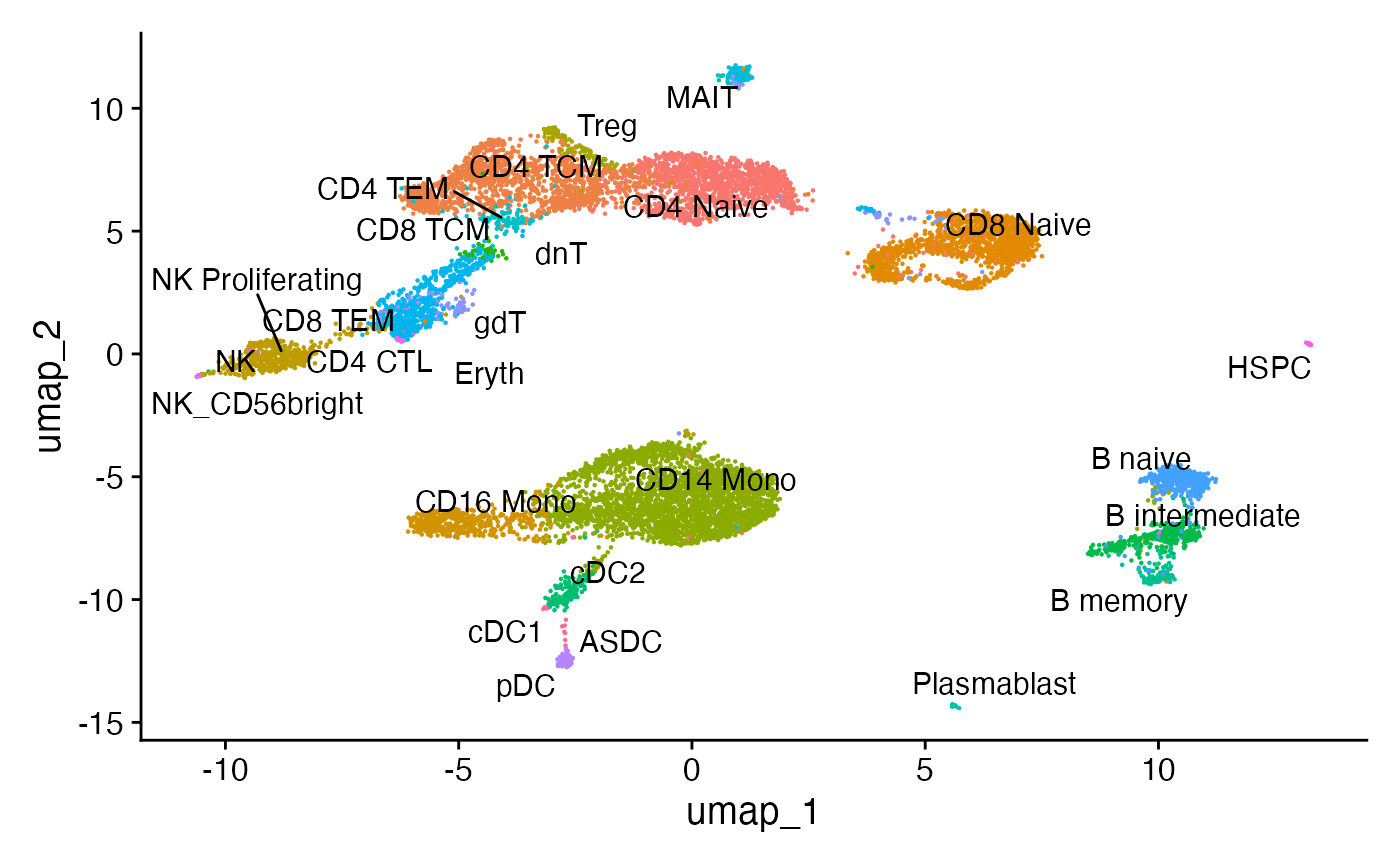
联合 UMAP 可视化
使用 Seurat v4 中的加权最近邻方法,我们可以计算代表基因表达和 DNA 可及性测量的UMAP图。
# build a joint neighbor graph using both assays
pbmc <- FindMultiModalNeighbors(
object = pbmc,
reduction.list = list("pca", "lsi"),
dims.list = list(1:50, 2:40),
modality.weight.name = "RNA.weight",
verbose = TRUE
)
# build a joint UMAP visualization
pbmc <- RunUMAP(
object = pbmc,
nn.name = "weighted.nn",
assay = "RNA",
verbose = TRUE
)
DimPlot(pbmc, label = TRUE, repel = TRUE, reduction = "umap") + NoLegend()
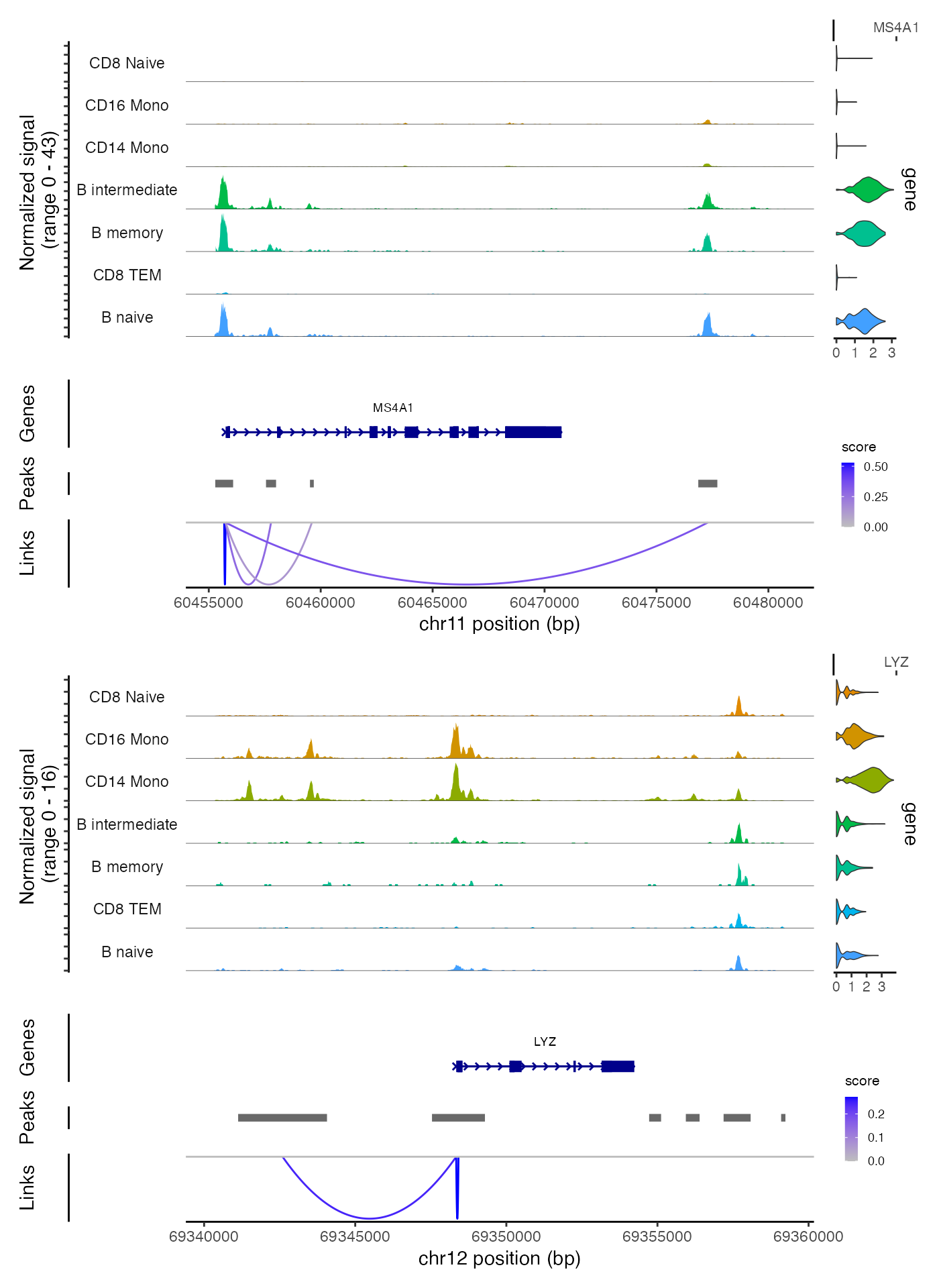
将峰与基因联系起来
为了找到可能调控每个基因的峰值集合,我们可以计算基因表达与其附近峰值可及性之间的相关性,并校正由于 GC 含量、整体可及性和峰值大小引起的偏差。
在整个基因组上执行这一步骤可能非常耗时,因此我们在这里以部分基因为例,展示峰-基因链接。通过省略 genes.use 参数,可以使用相同的函数来找到所有基因的链接:
DefaultAssay(pbmc) <- "ATAC"
# first compute the GC content for each peak
pbmc <- RegionStats(pbmc, genome = BSgenome.Hsapiens.UCSC.hg38)
# link peaks to genes
pbmc <- LinkPeaks(
object = pbmc,
peak.assay = "ATAC",
expression.assay = "SCT",
genes.use = c("LYZ", "MS4A1")
)
我们可以使用 CoveragePlot() 函数可视化这些链接,或者我们可以在交互式分析中使用 CoverageBrowser() 函数:
idents.plot <- c("B naive", "B intermediate", "B memory",
"CD14 Mono", "CD16 Mono", "CD8 TEM", "CD8 Naive")
p1 <- CoveragePlot(
object = pbmc,
region = "MS4A1",
features = "MS4A1",
expression.assay = "SCT",
idents = idents.plot,
extend.upstream = 500,
extend.downstream = 10000
)
p2 <- CoveragePlot(
object = pbmc,
region = "LYZ",
features = "LYZ",
expression.assay = "SCT",
idents = idents.plot,
extend.upstream = 8000,
extend.downstream = 5000
)
patchwork::wrap_plots(p1, p2, ncol = 1)
Source: https://stuartlab.org/signac/articles/pbmc_multiomic
本文由 mdnice 多平台发布
这篇关于单细胞|RNA-seq ATAC-seq 联合分析的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







