本文主要是介绍Signac|成年小鼠大脑 单细胞ATAC分析(1),希望对大家解决编程问题提供一定的参考价值,需要的开发者们随着小编来一起学习吧!
引言
在本教程中,我们将探讨由10x Genomics公司提供的成年小鼠大脑细胞的单细胞ATAC-seq数据集。本教程中使用的所有相关文件均可在10x Genomics官方网站上获取。
本教程复现了之前在人类外周血单核细胞(PBMC)的Signac入门教程中执行的命令。我们通过在不同的系统上进行相同的分析,来展示其性能以及对不同组织类型的适用性,并提供了一个来自不同物种的示例。
实战
首先,我们需要导入Signac、Seurat等一些用于分析小鼠数据的软件包。
library(Signac)
library(Seurat)
library(EnsDb.Mmusculus.v79)
library(ggplot2)
library(patchwork)
预处理工作流程
counts <- Read10X_h5("../vignette_data/atac_v1_adult_brain_fresh_5k_filtered_peak_bc_matrix.h5")
metadata <- read.csv(
file = "../vignette_data/atac_v1_adult_brain_fresh_5k_singlecell.csv",
header = TRUE,
row.names = 1
)
brain_assay <- CreateChromatinAssay(
counts = counts,
sep = c(":", "-"),
genome = "mm10",
fragments = '../vignette_data/atac_v1_adult_brain_fresh_5k_fragments.tsv.gz',
min.cells = 1
)
brain <- CreateSeuratObject(
counts = brain_assay,
assay = 'peaks',
project = 'ATAC',
meta.data = metadata
)
我们还可以向小鼠基因组的大脑对象添加基因注释。这将允许下游函数直接从对象中提取基因注释信息。
# extract gene annotations from EnsDb
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Mmusculus.v79)
# change to UCSC style since the data was mapped to hg19
seqlevels(annotations) <- paste0('chr', seqlevels(annotations))
genome(annotations) <- "mm10"
# add the gene information to the object
Annotation(brain) <- annotations
计算 QC 指标
接下来我们计算一些有用的细胞 QC 指标。
brain <- NucleosomeSignal(object = brain)
我们可以分析所有细胞的DNA片段长度的周期性变化,并根据细胞核小体信号的强弱进行分类。观察结果表明,那些在单核小体与无核小体比例上表现异常的细胞,呈现出与其他细胞不同的条带图谱。而其他细胞则显示出了一次成功的ATAC-seq实验所特有的典型模式。
brain$nucleosome_group <- ifelse(brain$nucleosome_signal > 4, 'NS > 4', 'NS < 4')
FragmentHistogram(object = brain, group.by = 'nucleosome_group', region = 'chr1-1-10000000')

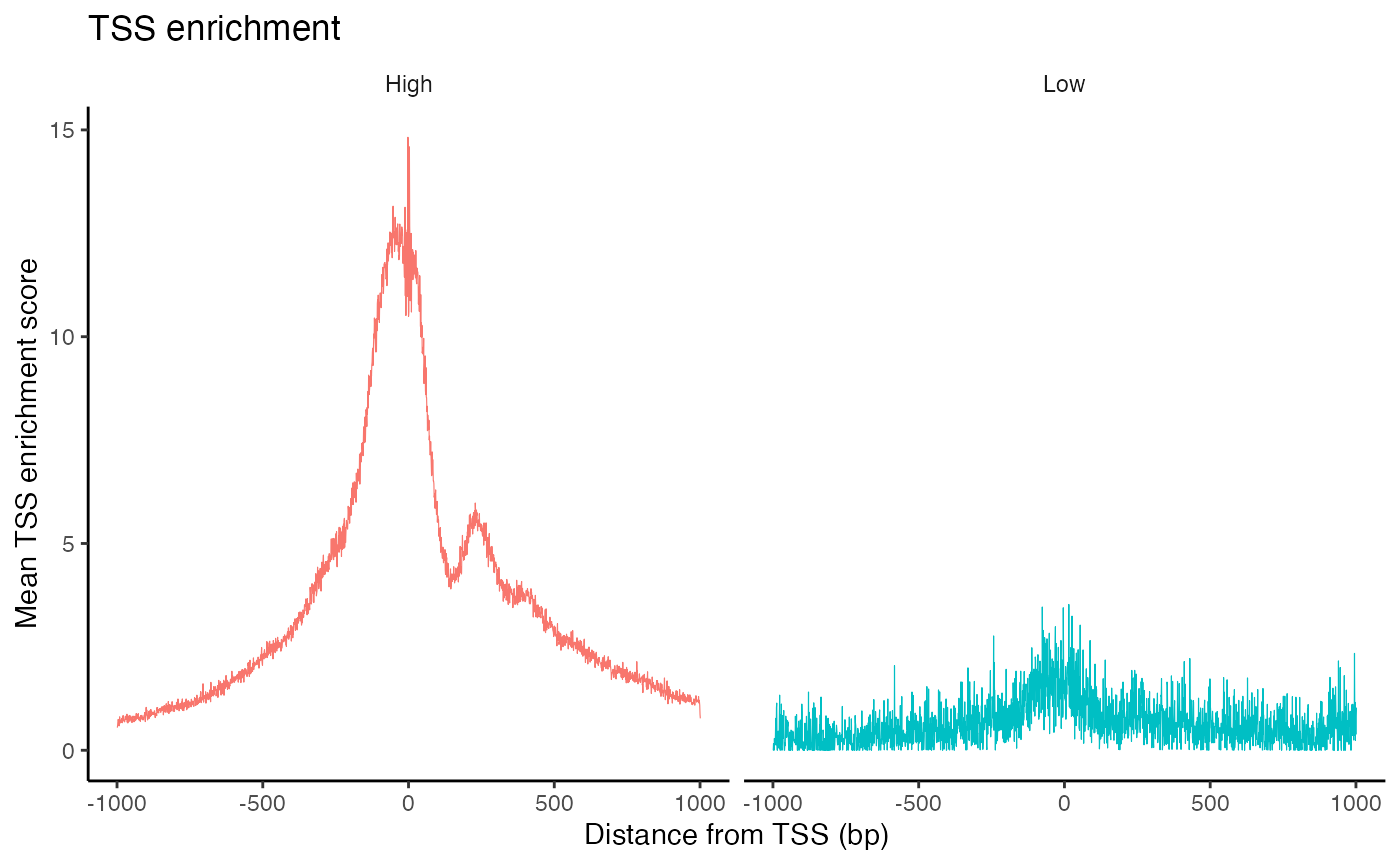
在ATAC-seq实验中,Tn5转座酶在转录起始位点(TSS)处的整合事件的富集程度,是一个关键的质量控制指标,用于评价Tn5的定位效率。ENCODE联盟将TSS富集分数定义为TSS周围Tn5整合位点的计数与这些位点在相邻区域计数的比率。在Signac软件包中,我们可以使用TSSEnrichment()函数来为每个细胞计算这一富集分数。
brain <- TSSEnrichment(brain, fast = FALSE)
brain$high.tss <- ifelse(brain$TSS.enrichment > 2, 'High', 'Low')
TSSPlot(brain, group.by = 'high.tss') + NoLegend()
brain$pct_reads_in_peaks <- brain$peak_region_fragments / brain$passed_filters * 100
brain$blacklist_ratio <- brain$blacklist_region_fragments / brain$peak_region_fragments
VlnPlot(
object = brain,
features = c('pct_reads_in_peaks', 'peak_region_fragments',
'TSS.enrichment', 'blacklist_ratio', 'nucleosome_signal'),
pt.size = 0.1,
ncol = 5
)

我们删除了这些 QC 指标异常值的细胞。
brain <- subset(
x = brain,
subset = peak_region_fragments > 3000 &
peak_region_fragments < 100000 &
pct_reads_in_peaks > 40 &
blacklist_ratio < 0.025 &
nucleosome_signal < 4 &
TSS.enrichment > 2
)
brain
## An object of class Seurat
## 157203 features across 3512 samples within 1 assay
## Active assay: peaks (157203 features, 0 variable features)
## 2 layers present: counts, data
归一化和线性降维
brain <- RunTFIDF(brain)
brain <- FindTopFeatures(brain, min.cutoff = 'q0')
brain <- RunSVD(object = brain)
在分析中,LSI(线性判别分析)的第一个主成分往往反映的是测序的深度(即技术层面的变异),而非生物学上的变异。如果确实如此,那么在后续的分析中应该将这一成分排除掉。为了判断是否存在这种情况,我们可以通过调用DepthCor()函数来计算每个LSI主成分与测序深度之间的相关性。
DepthCor(brain)

在这里,我们看到第一个 LSI 组件与细胞的计数总数之间存在非常强的相关性,因此我们将在没有该组件的情况下执行下游步骤。
非线性降维和聚类
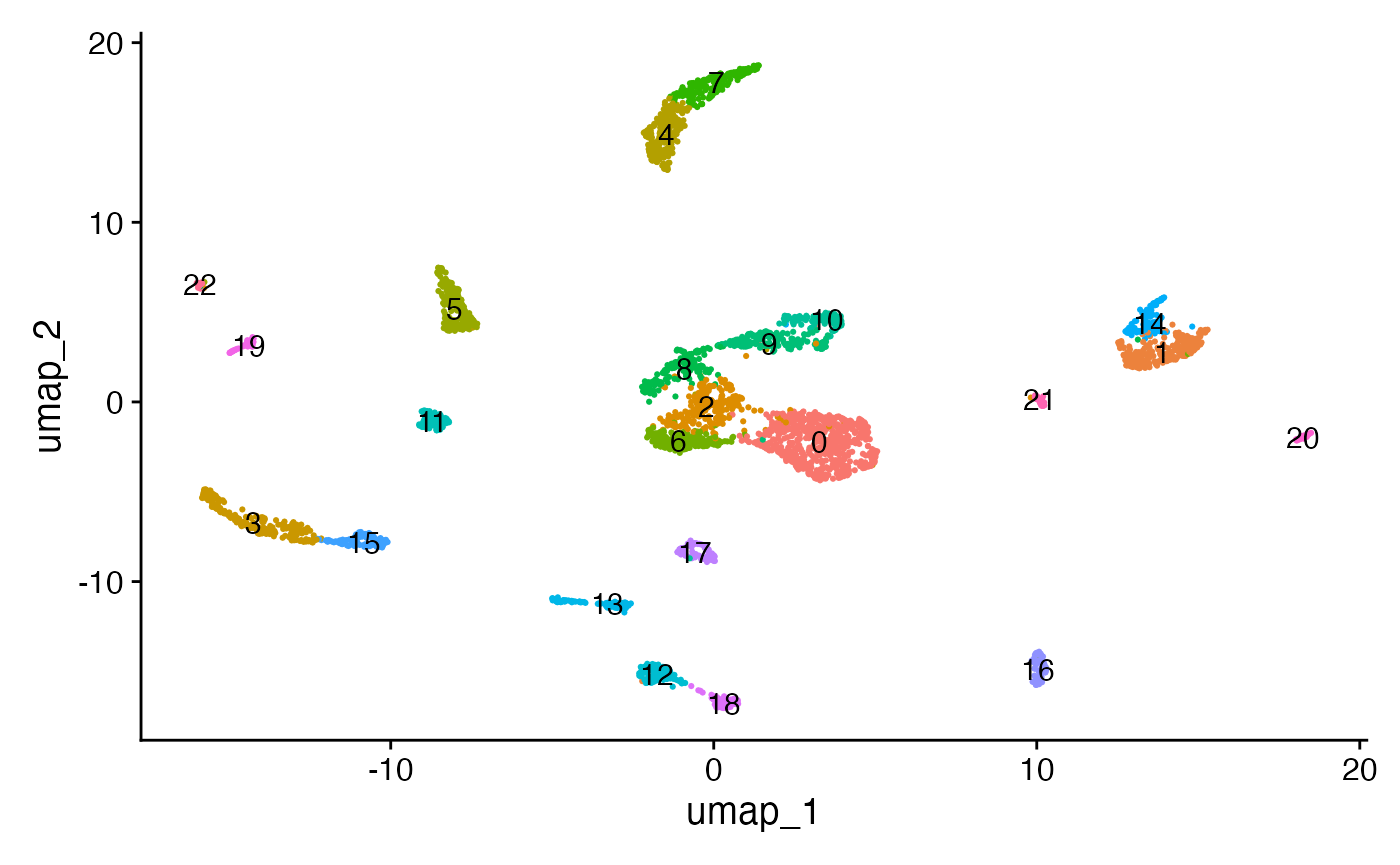
细胞数据已经被嵌入到一个低维度的空间里,我们可以采用单细胞RNA测序(scRNA-seq)数据常用的分析方法,执行基于图谱的聚类分析,并通过非线性降维技术来进行数据可视化。RunUMAP()、FindNeighbors()和FindClusters()这些功能均集成在Seurat软件包中。
brain <- RunUMAP(
object = brain,
reduction = 'lsi',
dims = 2:30
)
brain <- FindNeighbors(
object = brain,
reduction = 'lsi',
dims = 2:30
)
brain <- FindClusters(
object = brain,
algorithm = 3,
resolution = 1.2,
verbose = FALSE
)
DimPlot(object = brain, label = TRUE) + NoLegend()
本文由 mdnice 多平台发布
这篇关于Signac|成年小鼠大脑 单细胞ATAC分析(1)的文章就介绍到这儿,希望我们推荐的文章对编程师们有所帮助!







